
Kalp yetersizliği (KY) sendromunu tam olarak açıklayacak özgül tek bir patofizyolojik mekanizma ve bunu özetleyecek bir şema günümüze kadar bulunamamıştır. Önceleri klinisyenler kalp yetersizliğini böbrek kan akım anormalliklerine (kardiyorenal model) bağlı olarak fazlaca su ve tuz tutulması ve/veya kalbin pompalama kapasitesindeki anormallikler (kardiyosirkülatüer veya hemodinamik model) olarak görmüş olsalar da bu modellerden hiçbirisi bu sendromdaki kötüleşen seyri açıklayamamışlardır. Bu bölümde ağırlıklı olarak ejeksiyon fraksiyonu düşük kalp yetersizliğinde (KYdEF) alt da yatan moleküler ve hücresel değişiklikleri özelliklede bu sendromun ilerleyici yapısında baş rolü üstlenen nörohumaral aktivasyon ve sol ventrikül yeniden yapılanmasını (remodeling) vurgulayarak anlatılacaktır.
PATOGENEZ
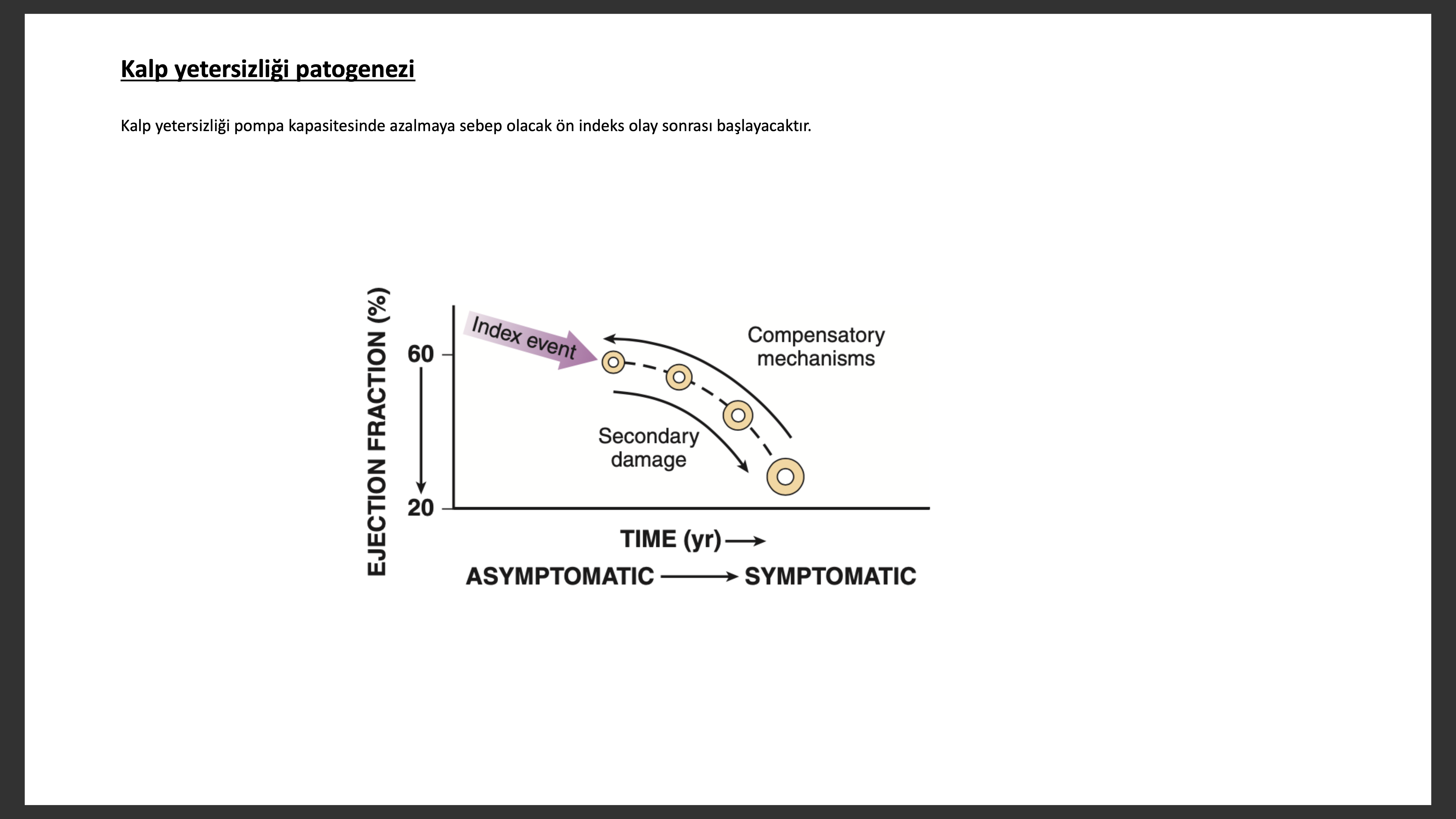
Kalp yetersizliği adaleyi zedeleyen ve miyositlerin fonksiyon kaybına yol açan veya miyokardın kuvvet oluşturmasını bozarak normal kasılmasını engelleyen indeks bir olay sonrası gelişen ilerleyici bir hastalıktır. Bu indeks olay miyokart infarktüsü (MI) gibi ani; sıvı, basınç yüklenmesi gibi veya herediter kardiyomiyopatiler gibi yavaş, sinsi seyirli olabilir. Başlangıçta olan olayın doğasından bağımsız olarak bu indeks olayların ortak noktası hepsinin bir şekilde kalbin pompa fonksiyonunu azaltmasıdır. Bu başlangıç kalp pompa fonksiyon azalması sonrasında çoğu hasta asemptomatik veya minimal semptomatik olacaktır. Çoğunlukla semptomlar bu fonksiyon bozukluğu gelişme indeks olayından belirgin bir süre sonra semptomatik olmaya başlarlar. Sol ventrikül fonksiyon bozukluğu gelişen hastaların neden asemptomatik oldukları kesin olarak söylenemez ise de potansiyel açıklamalardan ilki, bazı kompanse edici mekanizmaların kardiyak zedelenme veya debi düşüklüğü sırasında aktive olarak sol ventrikül fonksiyonunu fizyolojik/homeostatik sınırda tutması ve böylece hastaların fonksiyonel kapasitesinin korunması veya minimal azalması şeklindedir. Semptomatik kalp yetersizliğine doğru kalp yetersizliği ilerlerken nörohumoral ve sitokin sistemlerinin artmış aktivasyonu miyokart da sol ventrikül yeniden yapılanması (remodeling) denilen hedef doku değişiklikleri meydana getirir. Daha sonraları tartışılacağı gibi bu yeniden yapılanma hastanın nörohumoral durumundan bağımsız olarak kalp yetersizliğinde hastalığın ilerlemesine sebep olur.
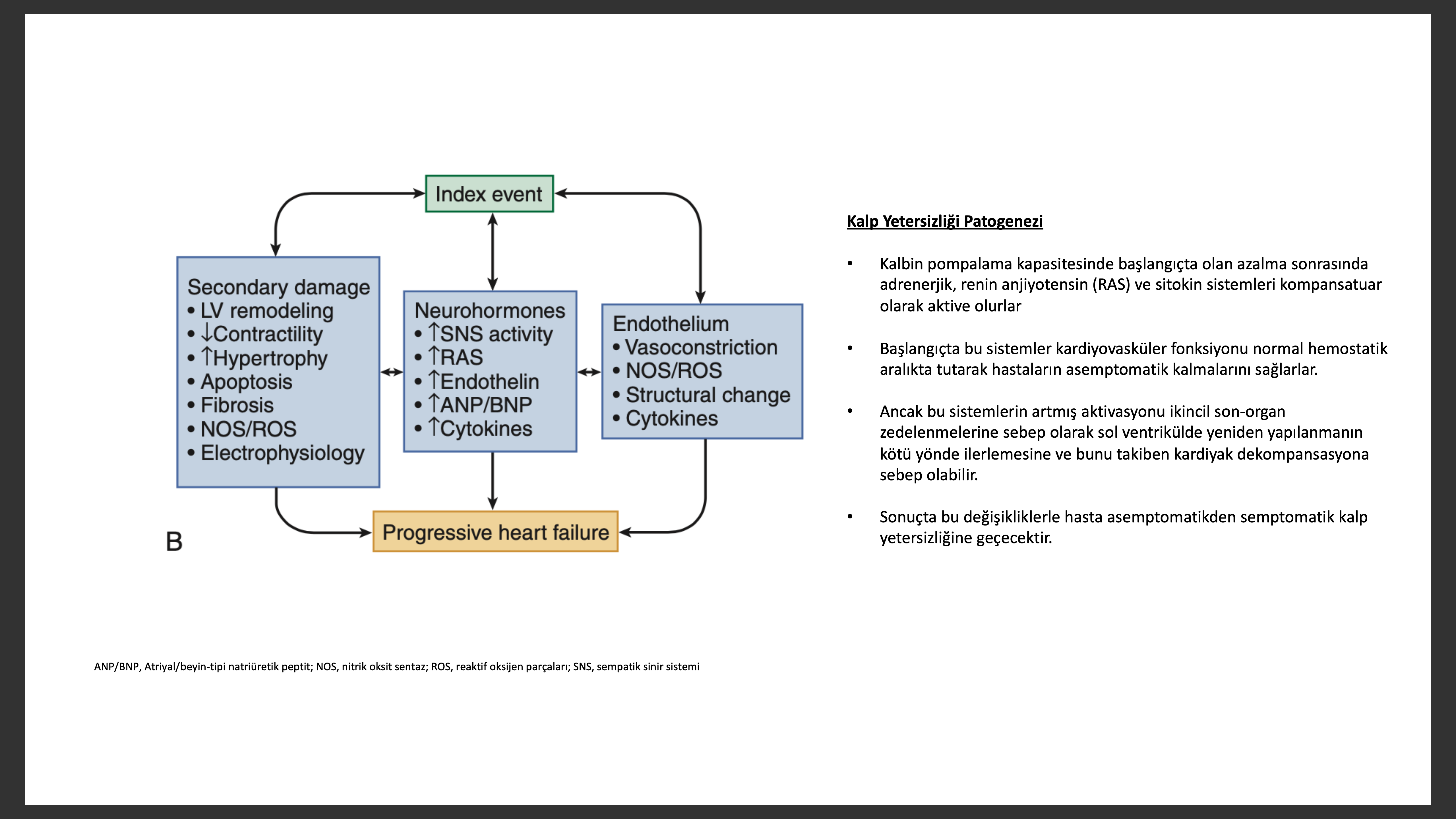
İLERLEYİEN BİR HASTALIK MODELİ; KALP YETERSİZLİĞİ
Nörohumoral Mekanizma
Klinik ve deneysel çalışmalardaki veriler biyolojik aktif moleküllerin fazlaca baskın olmasının kalp ve dolaşım üzerine olumsuz etkilerinin kalp yetersizliğinin ilerlemesine sebep olduğunu göstermektedir. Kompansatuar mekanizmaların en önemlileri adrenerjik sinir sistemi ve renin anjiyotensin sistemi (RAS), su ve tuz tutulumunu, periferik vazokonstriksiyonu ve kontraksiyonu arttırıp kardiyak debiyi arttırır. Aynı zamanda inflamatuar uyarıcı etki ile kardiyak onarım ve yeniden yapılanmaya (remodeling) sebep olur. Eskiden kullanılan nörohormon terimi özellikle kalp yetersizliğinde etki gösteren moleküllerin nöroendokrin sistemde üretilmesi ve hormon olarak etki etmesi sebebi ile kullanılmış. Nörohormon olarak isimlendirilmiş olmalarına rağmen norepinefrin (NE) ve anjiyotensin II (AgtII) direkt olarak miyozitler ile miyokart da üretilerek otokrin ve parakrin etkiler gösterir. Fazla salınımında bu molekül sisteminin tamamının kalp ve dolaşım üzerine olan olumsuz etkileri ve hastalık kötü seyri üzerine etkisi nörohumoral modelin benimsenmesinde etkili olmuştur.
Sempatik Sinir Sisteminin Aktivasyonu
Kalp yetersizliğinde debideki azalma kardiyovasküler homeostazı sağlamak için bazı kompansatuar sistemleri aktive etmektedir. Önemli adaptasyonlardan bir tanesi kalp yetersizliği erken devresinde olan sempatik sinin sisteminin (SSS) aktivasyonudur. KY de SSS aktivasyonu parasempatik tonun ortadan kalkması ile birliktedir. Başlangıçta kalp yetersizliğinde değişen otonomik etkinin arteriyel ve kardiyopulmoner baroreseptör inhibitör etkisinin ortadan kalkması şeklinde olduğu düşünülürken son zamanlardaki bulgular uyarıcı reflekslerinde bu otonomik dengesizliğe eklendiğini göstermektedir. Sağlıklı insanlarda karotid sinüs ve aortik ark “yüksek basınç” reseptörleri ve kardiyopulmoner “düşük basınç” mekanoreseptörleri santral sinir sistemine inhibitör sinyaller yollayarak sempatik sistem akışını kalpte ve periferik dolaşımda baskılar. Normalde karotid sinüs ve aortik ark “yüksek basınç” reseptörleri ve kardiyopulmoner “düşük basınç” mekanoreseptörleri sempatik sistemin en önemli inhibitör, nonbarorefleks periferik kemoreseptör ve adele metaboreseptörleri, sempatik sistemin en önemli uyarıcı reseptörleridir. Baroreseptör kalp hızı refleksi vagal uzvu, arteriyel baroreseptör afferent inhibitör etkisine de duyarlıdır. Sağlıklı insanlarda istirahat halinde sempatik salınım düşüktür ve kalp hızı değişkenliği (heart rate variability) yüksektir. Kalp yetersizliğinde baroreseptör ve mekanoreseptörlerde inhibitör etki azalır ve uyarıcı etki artar sonuçta sempatik sinir trafiği yoğunlaşıp, para sempatik sinir trafiği körelerek kalp hızı değişkenliği baskılanıp periferik vasküler direnç artacaktır.
Sempatik tonusdaki artış potent adrenerjik nörotransmitter NE in dolaşımdaki seviyesini artıracaktır. Dolaşımdaki NE artışı, adrenerjik sinir uçlarından salınma, plazmaya dökülme ve sinir uçlarından NE geri alınımı azalmasının kombinasyonu sonucudur. İleri kalp yetersizliği olanlarda dolaşımdaki dinlenme NE düzeyleri normal kişilere göre 2-3 kat daha fazladır. Kalp yetersizliği olan hastalarda kanda NE seviyelerinin yüksek olması mortalite göstergesidir. Normalde kalp NE i arteriyel kana atar, orta derecede kalp yetersizliği olan hastalarda koroner sinüs NE konsantrasyonunun kana göre daha yüksek olduğunun saptanması kalpte adrenerjik etkinin daha fazla olduğunu gösterir. Ancak kalp yetersizliği ilerledikçe miyokardiyal NE konsantrasyonunda belirgin azalma olur. Ciddi kalp yetersizliğinde kalpte NE azalmasının mekanizması belli değildir, kalp yetersizliğinde kardiyak adrenerjik sinirlerde uzamış adrenerjik aktivasyona bağlı “yorulma” fenomenine bağlı olabilir. Ek olarak NE sentezinin hız sınırlayan enzimi miyokardiyal tirozin hidroksilaz aktivitesi kalpte azalmıştır. Kardiyomiyopatili hastalarda radiyofarmakolojik olarak adrenerjik sinir sonlarınca alınan iyodine 131 ile işaretlenen metaidobenzilguanidine in normal olarak alınmaması NE geri alınımının da etkilenmiş olduğunu düşündürmektedir.
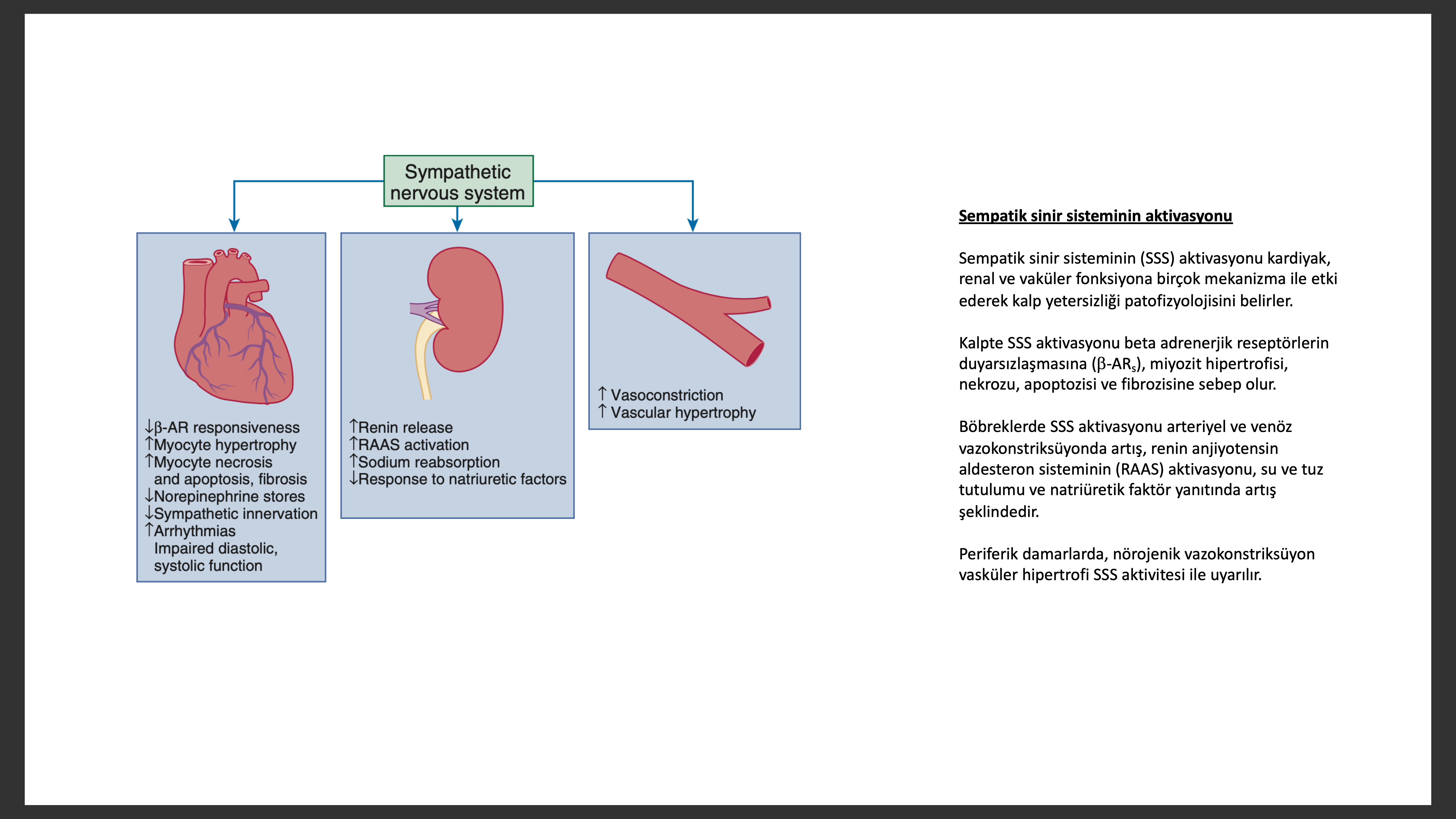
Beta1 -adrenerjik reseptör ile artan sempatik aktivasyon kalp hızı ve miyokardiyal kasılma kuvvetini artırarak kardiyak debiyi artırır. Ek olarak artmış adrenerjik sinir sistemi aktivitesi miyokardiyal alfa1-adrenerjik reseptörleri uyarır, sonucunda orta derecede pozitif inotropi ve periferik arterlerde vazokonstriksiyonu olur. NE kontraksiyon ve relaksayonu artırması ve kan basıncını korumasına rağmen miyokardiyal oksijen ihtiyacını da artıracaktır, bu durum özellikle miyokarta oksijen getirilmesinin sınırlı olduğu durumlarda iskemiyi artıra bilir. Santral sinir siteminde artan adrenerjik uyarı özellikle miyokardiyal iskemi olan durumlarda ventriküler taşikardileri ve ani kardiyak ölümü tetikleye bilir. SSS aktivasyonu kısa dönemde destek sağlamakla birlikte uzun dönemde adaptasyon bozukluğuna sebep olabilir. Dahası gün geçtikçe daha fazla delil sempatik aktivasyonun zararlı etkisine ek olarak parasempatik etkinin ortadan kalkmasının da kalp yetersizliği patogenezinde etkili olabileceğini göstermektedir. Parasempatik etkinin ortadan kalkmasının nitrik oksit (NO) seviyelerinde azalma, inflamasyonda artma, sempatik aktiviteyi arttırma, ve sol ventrikül yeniden yapılanmasını daha kötü hale getirme gibi etkileri olduğu düşünülmektedir. Vagal siniri uyararak yapılan bazı çalışmalar birincil son noktalarında bu sonuçları göstermemiş olsa dahi ikincil son noktalarda bu etkiyi göstermişlerdir.
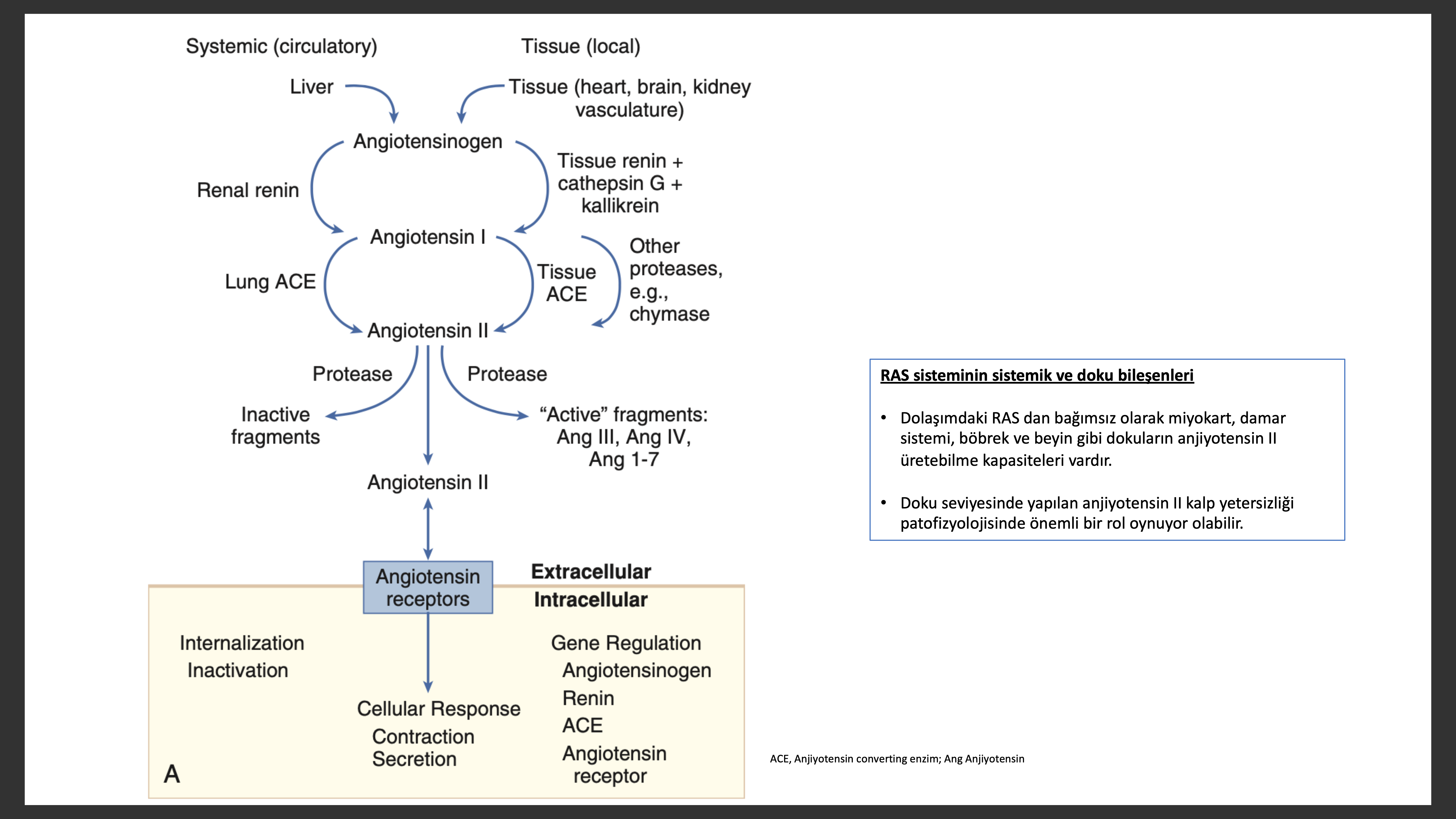
Renin -Anjiyotensin Sisteminin Aktivasyonu
SSS oranla KY de RAS daha geç aktive hale gelir . Kalp yetersizliğinde RAS aktivasyonundaki görüş renal hipoperfüzyon, distal tübülde makule densaya gelen filtre sodyum miktarı azalması ve artmış SSS aktivitesinin böbrek jukstamedüller aparatından renin salınımını arttırması şeklinde olduğu yönündedir. Renin karaciğerde sentez edilen dolaşımdaki anjiyotensinojenden dört aminoasit kopararak biyolojik olarak aktif olmayan dekapeptit anjiyotensin I i oluşturur. Anjiyotensin converting enzim (ACE) anjiyotensin I den iki aminoasit kopararak biyolojik olarak aktif oktapeptit (1-8) anjiyotensin II yi oluşturur Vücuttaki ACE nin çoğunluğu (%90) dokuda bulunur; kalan % 10 is kalp ve damar duvarında intersisyumunda çözünür olarak (membrana bağlı olmadan) bulunur. Doku ACE sinin kalp yetersizliğindeki önemi, ACE ulak RNA (mRNA),ACE-bağlama kısımları ve ACE aktivitesinin çıkarılmış kalplerde artmış olmasıdır. Anjiyotensin II reninden bağımsız yollar ile anjiyotensinojeni anjiyotensin I e kallikrein ve katepsin G enzim yolu ile çevirerek elde edebilir. Dokuda anjiyotensin II yapımı da ACE den bağımsız olarak chymaze aktivasyonu ile yapıla bilir. Bu ikinci yol özellikle ACE kullanımı ile kalpte renin ve anjiyotensin I artığında miyokart için önem taşıya bilir.
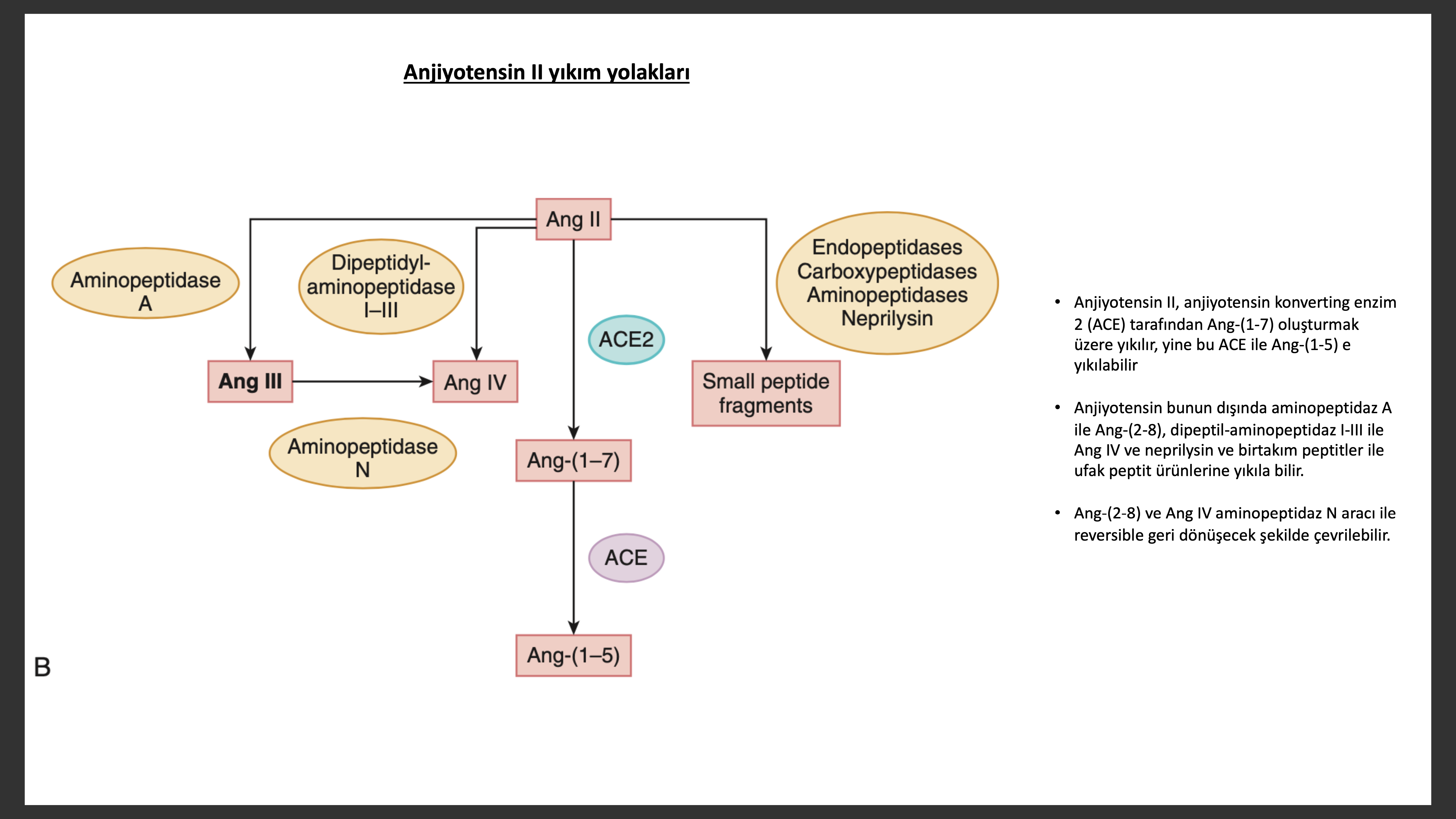
Anjiyotensinin kendisi proteolize giderek üç adet biyolojik aktif fragman ; anjiyotensin III (2-8), anjiyotensin IV (3-8) ve anjiyotensin 1-7 oluştura bilir.
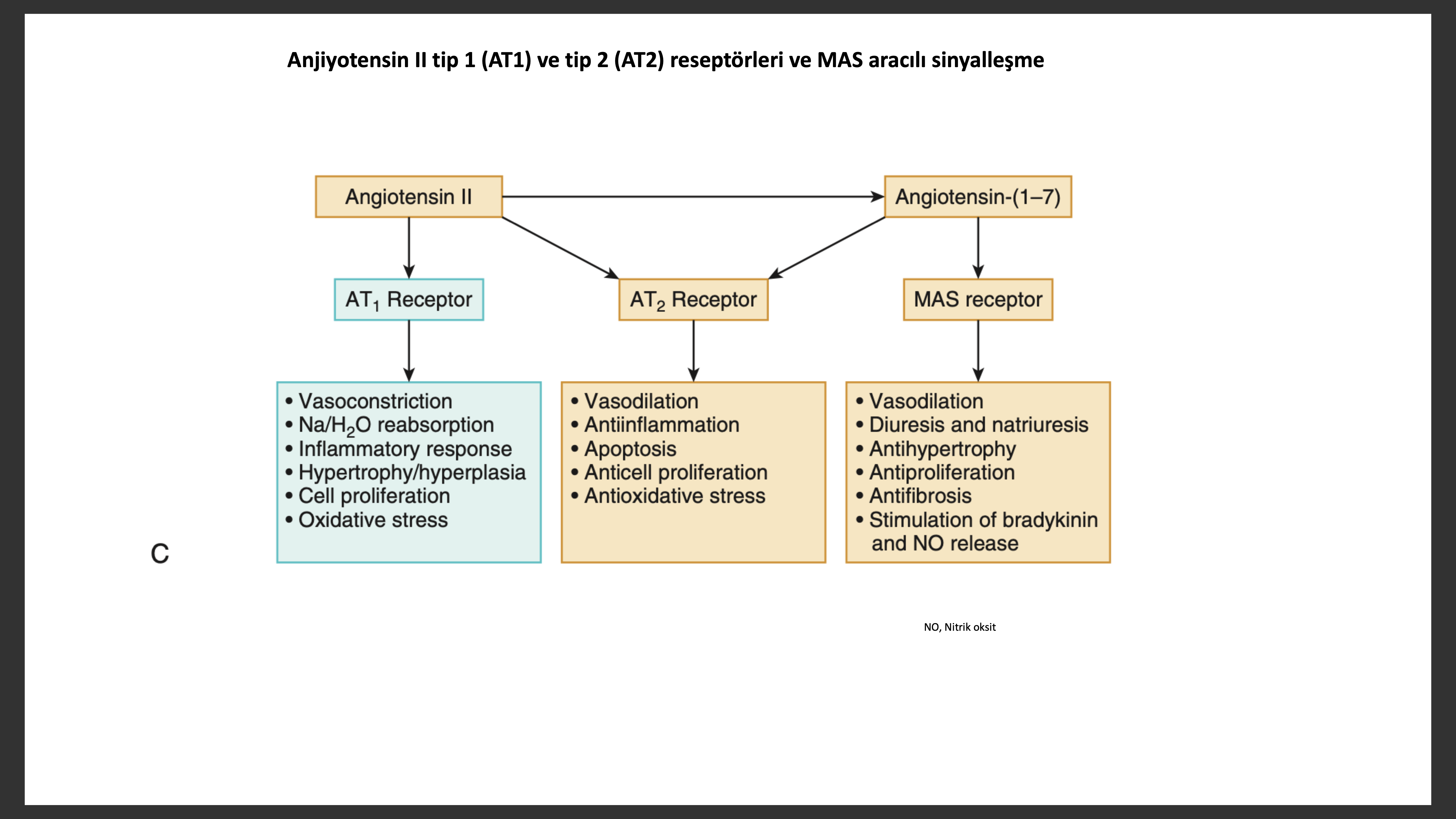
Anjiyotensin II etkisini iki adet G protein-çiftleşen reseptör (GPCRs) ile bağlanarak gösterir. Anjiyotensin tip 1 (AT1) ve anjiyotensin tip 2 (AT2) reseptörleri. Damar sisteminde baskın olan AT1 reseptörüdür. İnsan kalbinde hem AT1 hem de AT2reseptörü olmasına rağmen, AT2 reseptörü 2:1 oranında daha baskındır. Daha çok hücrede sinirlerin dağıldığı yerlerde hakimken daha çok fibroblastlar ve interstityum da bulunur . AT1 reseptör aktivasyonu vazokonstriksiyon, hücre büyümesi, aldesteron ve katekolamin salınımı yaparken AT reseptör aktivasyonu vazodilatasyon, hücre büyüme inhibisyonu, natriürez ve bradikinin salınımı yapmaktadır. Çalışmalar yetersizlik olan kalplerde AT1 reseptör ve mRNA seviyelerinin azaldığını, halbuki AT2 reseptör yoğunluğunun artığını veya değişmediğini, AT1/ AT2 oranının azaldığını göstermektedir. MAS reseptörü bir GPCR dır ve primer olarak beyin ve testiküller de bulunurken kalpte de miktarı yüksektir.
Anjiyotensin II nin kısa süreli dolaşım hemostazını sağlamada önemli birkaç etkisi vardır. Anjiyotensinin adapte olarak baskın olması kalpte, böbrekte ve diğer organlarda fibrozise sebep olacaktır. Anjiyotensin II sempatik sinir sonlarından NE salınımını artırarak nörohumaral aktivasyonun daha kötü bir almasına ve adrenal kortekste zona glomerulozayı uyararak aldesteron salınmasına sebep olacaktır. Anjiotensin II gibi aldesteron da nefronun distal kesimlerinden sodyum geri alınımını potasyuma karşılık artırarak kısa süreli hemodinamik destek sağlayacaktır. Aldesteronun baskın olarak salınması vasküler sistemde hipertrofi ve fibrozis gibi etkiler yaparak vasküler kompliansın azalması ve ventrikül sertliğinin artması gibi zararlı katkıda bulunacaktır. Ayrıca aldesteron endotel hücre ve baroreseptör disfonksiyonu , NE geri alım inhibisyonu yaparak kalp yetersizliğinin kötüleşmesine sebep olacaktır. Kardiyovasküler sistemde aldesteronun etkisi oksitadif stress ve ona bağlı hedef dokuda inflamasyon oluşması sonucu gibi gözükmektedir.
Anjiyotensin III(2-8), anjiyotensin IV (3-8) ve Anjiyotensin 1-7 nin kalp yetersizliğinde etki mekanizması bilinmemekle birlikte deneysel çalışmalar anjiyotensin 1-7 nin anjiyotensin II ile etkileşerek etkisini azaltıp, sol ventrikül yeniden yapılanmasını azalttığını göstermektedir.
Tersine anjiyotensin III zona glomeruloza yı uyararak adrenal bezlerden aldesteron salınımını arttırmakta ve distal toplayıcı kanal sodyum geri alınımını arttırmaktadır. Ayrıca anjiyotensin III ün beyinden vazopresin salınımını artırarak, distal böbrek toplayıcı kanallarından su tutulumunu artırdığı düşünülmektedir. Anjiyotensin III beyinde kardiyak sempatik hiper aktiviteye sebep olarak MI sonrası sol ventrikül yeniden yapılanmasına sebep olduğu düşünülmektedir.
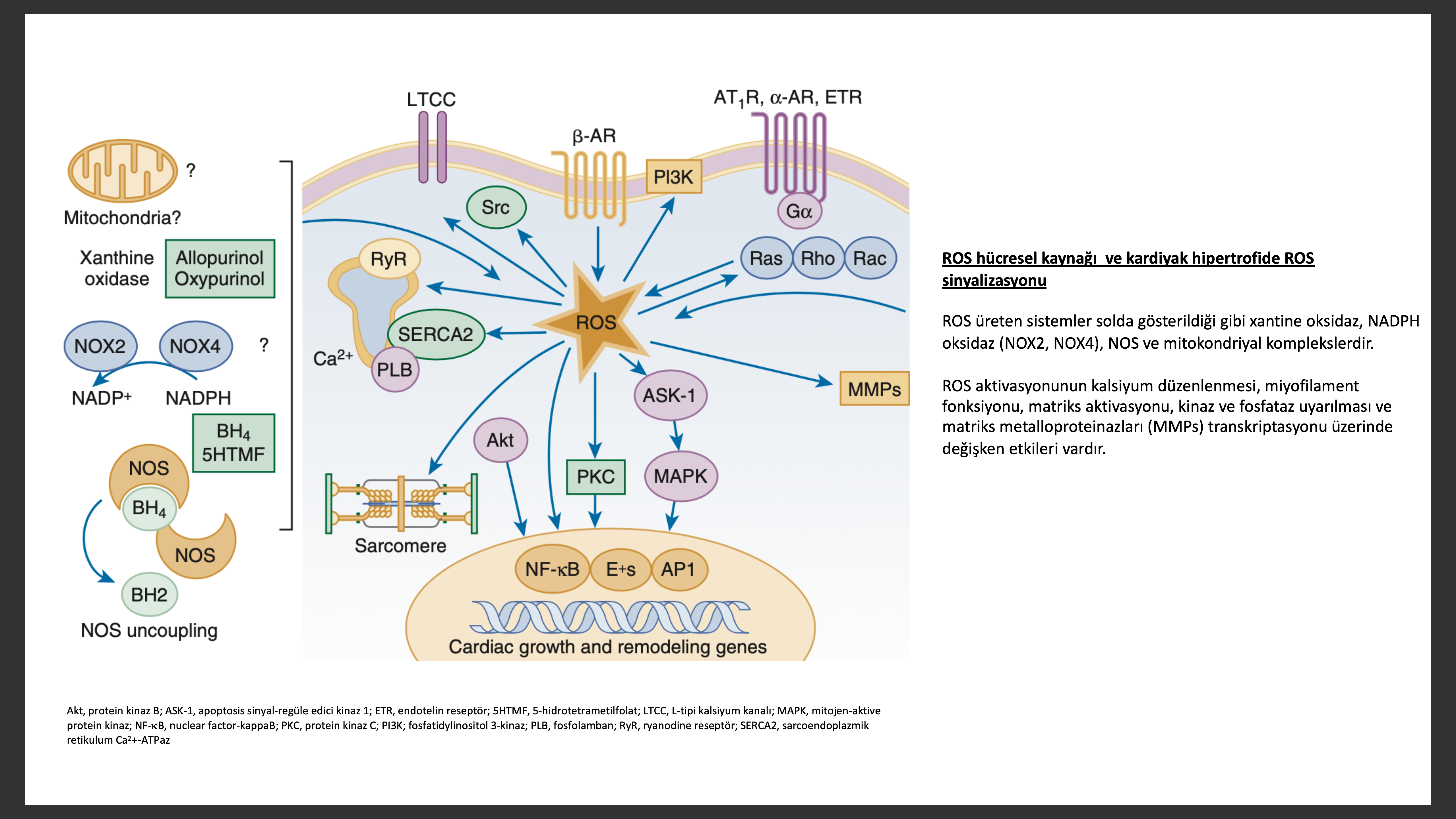
Oksitadif Stress;
Reaktif oksijen türleri (ROS) aerobik metabolizmanın normal ürünleridir. Kalpte mitokondri, xantine oksidaz, nikotinamide adenin dinükleotit fosfat (NADPH) oksidaz potansiyel ROS kaynaklarıdır. ROS, miyokardiyal eksitasyon-kontraksiyon çiftleşmesi için gerekli iyon kanalları, sarkoplazmik retikulum (SR), kalsiyum salınım kanalları, miyoflaman gibi bir çok intrasellüler protein aktivitesini ve miyosit büyümesi için eşleşen sinyal ulaklarını uyarır. “ Oksidatif stress” ROS yapımı antioksidan defans sisteminin tamponlayıcı kapasitesini aştığı zaman hücre içi fazla ROS birikince olur. Eldeki deliller kalp yetersizliğinde sistematik ve miyokart da oksidatif stresin arttığı yönünde. Kalpteki oksidatif stress miyokardın mekanik gerilimine , nörohumoral uyarıya(anjiyotensin II, alfa-adrenerjik agonistler, endotelin-1 [ET-1]) veya inflamatuar sitokinlere ( tümör nekrozis factor [TNF], interlökin[IL]-1) bağlı azalmış, antioksidan kapasite ve artmış ROS miktarına bağlı olabilir. Kalp yetersizliği deneysel modellerinde miyositler de mitokondri kaynaklı ROS fazlalığı gösterilmiştir. Hızlı ventrikül uyarımı ile kalp yetersizliği tetiklenmiş köpek deneylerinde ve son dönem kalp yetmezliği olan hastalarda xantin oksidaz miktarı ve aktivitesi artmıştır. Hem deneysel hem de klinik kalp yetersizliği çalışmalarında miyokardiyal NADPH oksidaz miktarı ve aktivitesi artmıştır. ROS ,kültürlenen kardiyak miyozitler de miyozit hipertrofisini, fetal gen program yeniden ortaya çıkmasını ve apoptozisi aktive eder. Yine ROS firoblast proliferasyonu, kollajen sentezi ve matriks metalloproteinaz (MMP) artışına katkıda bulunur. ROS yine NO bulunuşunu azaltarak kalp yetersizliğinde periferik damar ağını azaltır. Bütün bu bulgular kalp yetersizliği olan hastalarda ROS azaltılmasının tedavi değeri olabileceğini düşündürmektedir. Ancak hiperürisemik hastalarda allopurinol ile xantine oksidazın azaltılarak oksidatif stressin azaltıldığı hastalarda yeni bir çalışmada klinik statü ve kardiyak fonksiyonlarda fayda gösterilemedi. Anjiyotensinden bağımsız olarak spirinolaktonun klinik çalışmalarda New York kalp cemiyeti (NYHA) klass II- IV semptomatik sistolik kalp yetersizliği olan ve MI sonrası, volüm yükü veya elektrolit durumundan bağımsız olarak hayatta kalımı artırdığı gösterilmiştir.
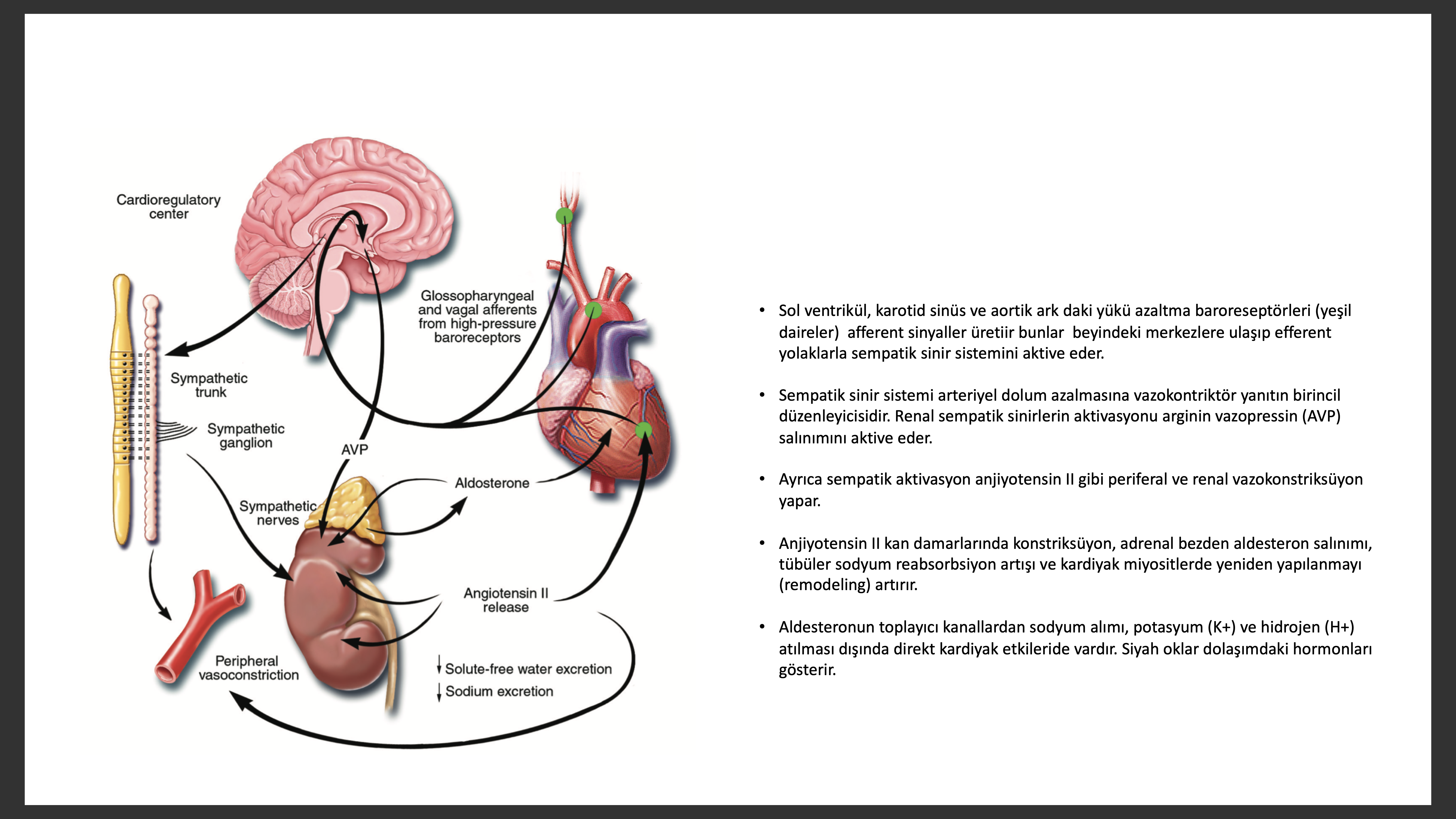
Renal Fonksiyon Nörohumoral Değişimi
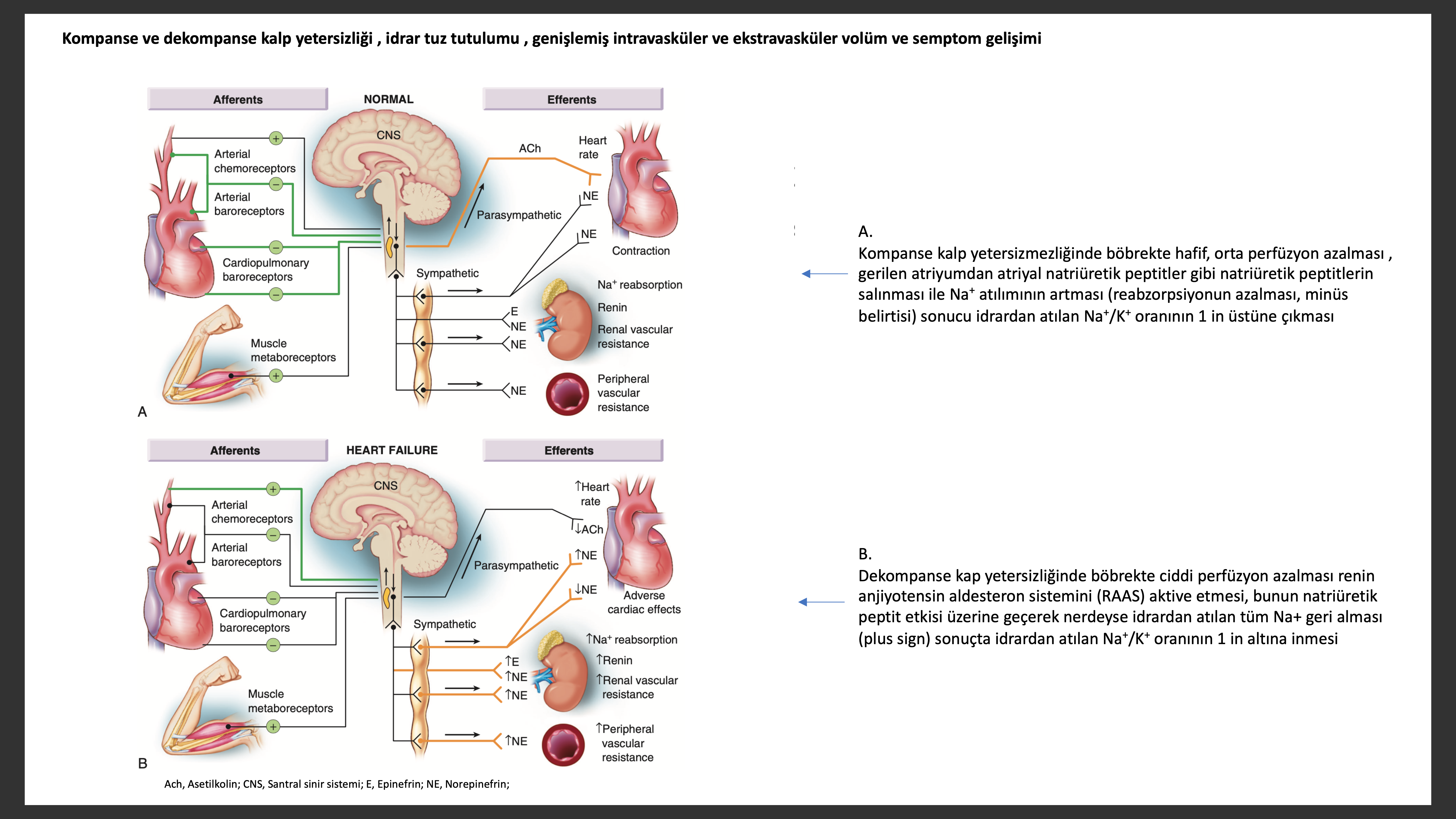
İlerleyen kalp yetmezliğinin imza niteliğindeki özelliği böbreklerden su ve tuz tutulmasını artırmasıdır. Geleneksel olarak kardiyak debinin “ileri” doğru azalması ile böbrek perfüzyonu azalması tuz tutulumu veya “geriye” doğru yetersizlik ile kanın göllenmesi, venöz basıncın artarak su ve tuzun intravaskülerden ekstravasküler bölgeye transudasyonu teorileri ileri sürülmektedir. Bu mekanizmalar yeterli arteriyel kan volümünün kalp yetersizliğinde kan volüm genişlemesine rağmen azalmış olması, vasküler yataktaki baroreseptörlerin bu uygunsuz kan akımını ,akut kan kaybına olan homeostatik yanıta benzer şekilde sezerek bir takım kompanse edici nörohumoral adaptasyon ile yanıt vermesi şeklinde açıklanır. Yetersiz debi veya kan akımının yeni düzenlenmesi, sol ventrikül, aortik ark, karotit sinüs ve renal afferent arteriyollerce algılanır. Arteriyel veya kardiyopulmoner refleklere gelen inhibitör uyarıların kaybı SSS ve RAS sistemini aktive eder. Karotit baroreseptörleri aktive ederek sempatik aktivasyonu azaltan ve vagal tonusu artıran implante edilebilir bir cihaz semptomatik kalp yetersizliği hastalarında hayat kalitesini ve efor kapasitesini artırmıştır. Halen devam eden BeAT-HF (Barostim Theraphy for Heart Failure) çalışması baroreseptör aktivasyon tedavisinin çalışma sonunda kariyovasküler ve kalp yetersizliği mortalitesinin çalışma sonlanmasında (etkinlik son noktası) ve 6 ayda majör nörolojik ve kardiyovasküler son noktalar hakkında bize bilgi verecektir.
Na+ birikiminin birincil olarak böbrekteki bir anormallikten kaynaklandığını düşündürecek yeterli delil olmamasına rağmen kalp yetersizliği ilerledikçe böbrekte olan ikincil bazı değişikliklerin sıvı yüklenmesine sebep olduğunu düşündürecek oldukça bulgu vardır. Kalp yetersizliğinde sıvı yüklenmesi ikinci olarak olur pek çok faktör etkilidir. Örneğin SSS ve RAS aktivasyonu, renal perfüzyonu azalması, ayrıca nefrondan renal tubüler sodyum ve su tekrar geri alınması bu ikicil etkilerdir. Renal sempatik stimulasyon ile hipofiz bezi posteriorundan arginin vazopressin (AVP) nonosmatik salınımı sıvı atılımını azaltacak, periferik vazokonstriksüyonun daha kötü hale gelmesine sebep olacak aynı zamanda endotelin (ET) salınımını artıracaktır. Artan renal venöz basınç, renal interstisyel basınç artışına sebep olarak tubular hasar ve renal fibrozise sebep olur.
Arginin Vazopressin
AVP serbest su temizleme ve plazma osmolarite ayarlanmasında önemli rol oynayan hipofiz bezi hormonudur. Normal koşullarda, AVP plazma ozmolaritesinin arttığı durumlarda salınarak toplayıcı kanaldan suyun geri alınmasını sağlar. Dolaşımdaki AVP çoğunlukla kalp yetersizlik hastalarda yüksektir ve osmolarite düzeldikten sonra dahi yüksek kalır (nonosmotik salınım) ve kalp yetersizliğindeki hiponatremi gelişmesinde rol alır. AVP nin hücresel etkileri V1a, V2a ve V2 denen üç tip reseptör ile olur. V1a en yaygın bulunan vasküler düz adalede bulunan subtipdir. V1b dağılımı sınırlıdır daha çok santral sinir sisteminde bulunur. V2 reseptörleri birincil olarak renal toplayıcı kanalların epitelyal hücrelerinde ve kalın çıkan bölümde yer alır. AVP reseptörleri GPCRs üyeleridir.V1a reseptörleri vazokonstriksiyon, platelet agregasyonu ve miyokardiyal büyüme faktörünü uyarırken, V1b hipofiz ön kısmından adrenokortikotropik (ACTH) hormon salınımını uyarır. V2 reseptörü adenil siklazı uyarıp apikal membran su kanalı-içeren veziküllere su geçişini artırarak diüretik etki yapar. Veziküllerde hazır fonksiyonel su kanalları aquaporinler bulunur, apikal membranda lokalize olan kanalların V2 uyarısına bağlı olarak su geçirgenliği artar ve su retansiyonu olur. Vazopresin reseptör antagonisti olan vaptanlar, V1a (relcovaptan) veya V2 (tolvaptan, lixivaptan) selektif olarak veya selektif olmadan V1a/V2 aktivitesi (conivaptan) vücut ağırlığı ve hiponatremiyi azaltır.
Artmış renal sempatik aktivite böbreklerde renin üretimini arttırarak ektrasellüler volüm artmış olmasına rağmen RAS aktivitesinde artışa sebep olur. Anjiyotensin II direkt proksimal tübüler etki, distal tübülden sodyum geri alınmasını artıran aldesteronu aktive etmesi gibi birçok renal mekanizma ile tuz ve su tutulumunu artırır. Ayrıca anjiyotensin II beyindeki susuzluk merkezini uyararak AVP ve aldesteron salınımını artırarak su ve tuz salınım düzenlenmesinde olumsuzluklar yapar.
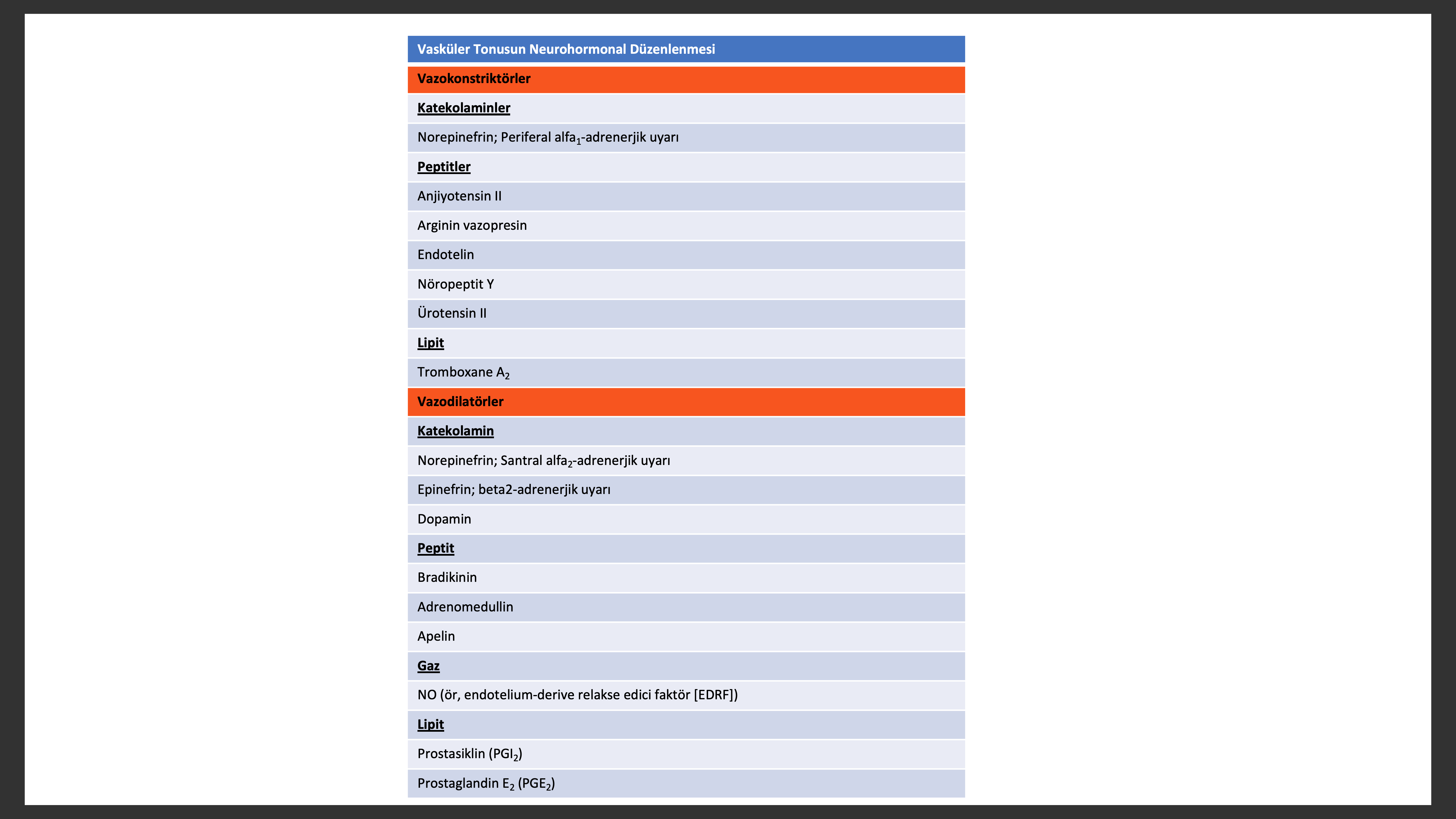 Kalp yetersizliğinde vazokonstriktör nörohormonların zararlı etkilerini ortadan kaldırmak için pek çok ters düzenleyici nörohormonal sistem de aktive olur. Kalp yetersizliğinde vazodilatör prostaglandin metabolitleri prostoglandin E2 (PGE2) ve prostasiklin (PGI2) miktarı artmıştır. PGE2 renal sodyum atılımını artırır ve AVP anti diüretik etkisini azaltır. Periferik vazokonstriksüyonun en kuvvetli uyaranı sempatik aktivasyon ve kuvvetli vazokonstriktör norepinefrin salınımıdır. Diğer dolaşımdaki homeostazı sağlayan vazokonstriktörler anjiyotensin II, ET, nöropeptit Y, ürotensin II, tromboksane A2 ve AVP dir.
Kalp yetersizliğinde vazokonstriktör nörohormonların zararlı etkilerini ortadan kaldırmak için pek çok ters düzenleyici nörohormonal sistem de aktive olur. Kalp yetersizliğinde vazodilatör prostaglandin metabolitleri prostoglandin E2 (PGE2) ve prostasiklin (PGI2) miktarı artmıştır. PGE2 renal sodyum atılımını artırır ve AVP anti diüretik etkisini azaltır. Periferik vazokonstriksüyonun en kuvvetli uyaranı sempatik aktivasyon ve kuvvetli vazokonstriktör norepinefrin salınımıdır. Diğer dolaşımdaki homeostazı sağlayan vazokonstriktörler anjiyotensin II, ET, nöropeptit Y, ürotensin II, tromboksane A2 ve AVP dir.
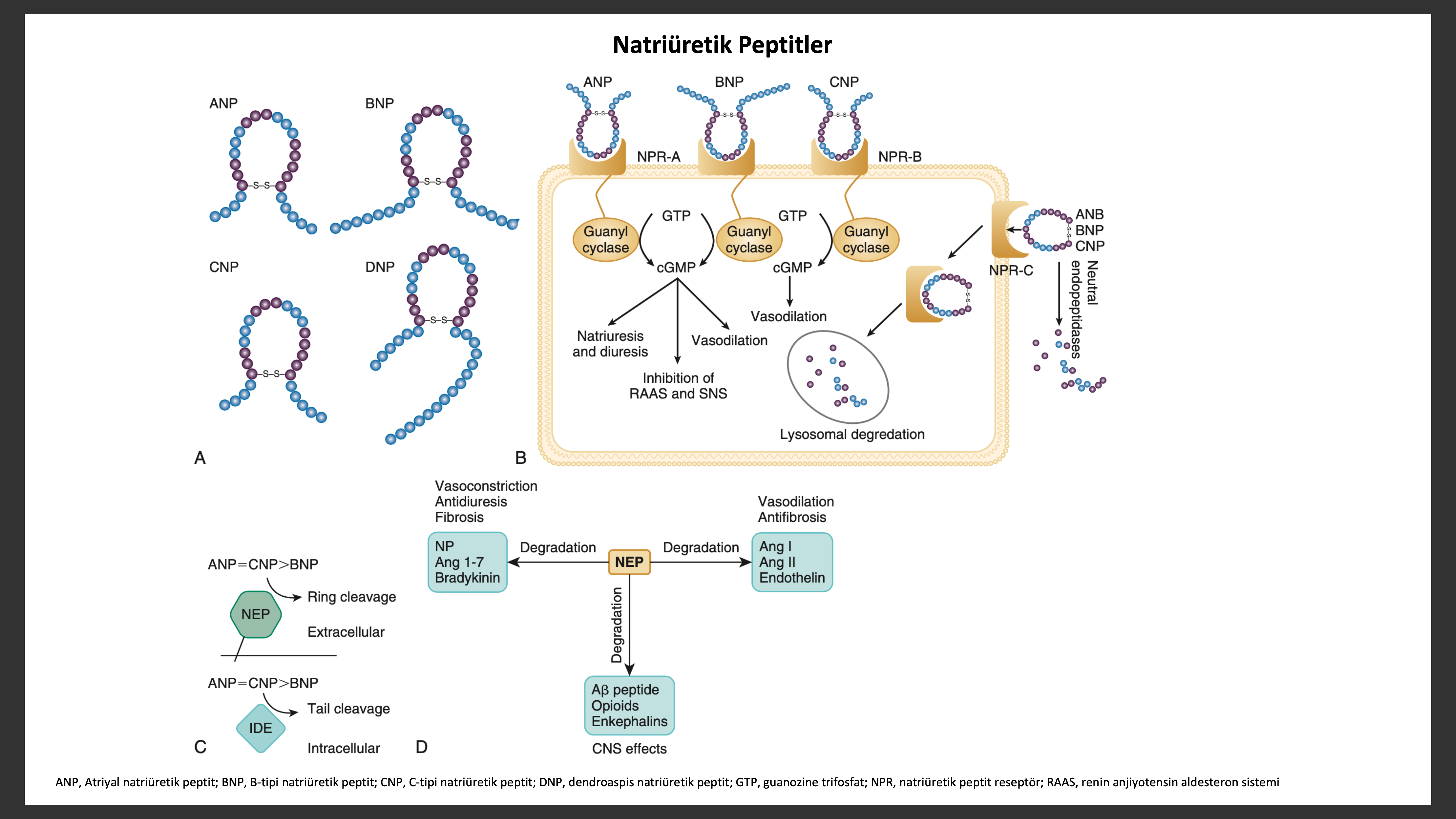
Kalp yetersizliğinde ters yönde düzenleyici bir başka nörohormon sistemi, atriyal natriüretik peptit (ANP) ve beyin (B-tipi) natriüretik peptit (BNP) den oluşan natriüretik peptitlerdir. Fizyolojik koşullarda ANP ve BNP, fazla tuz alımı sonrası miyokardiyal ve atriyal gerilime bağlı natriüretik hormon olarak salınırlar. Bir kere salınınca bu peptitler böbrek ve periferik dolaşımda sodyum ve su atılımını artırıp, renin ve aldesteron salınımını azaltarak kalbin yükünü azaltır. Sırasında RAS aktivasyonu sırasında ANP ve BNP ters düzenleyici görev yaparak sodyum ve su homeostazını sağlarlar.Çok iyi bilinmeyen sebeplerden dolayı ilerleyen kalp yetersizliğinde natriüretik peptitlerin renal etkileri körleşir ve RAS rakipsiz kalır. Düşük renal perfüzyonu basıncı, natriüretik peptit moleküler form alternasyonu ve defekti, natriüretik peptit reseptör azalması körelmenin potansiyel sebepleridir.
Natriüretik Peptitler
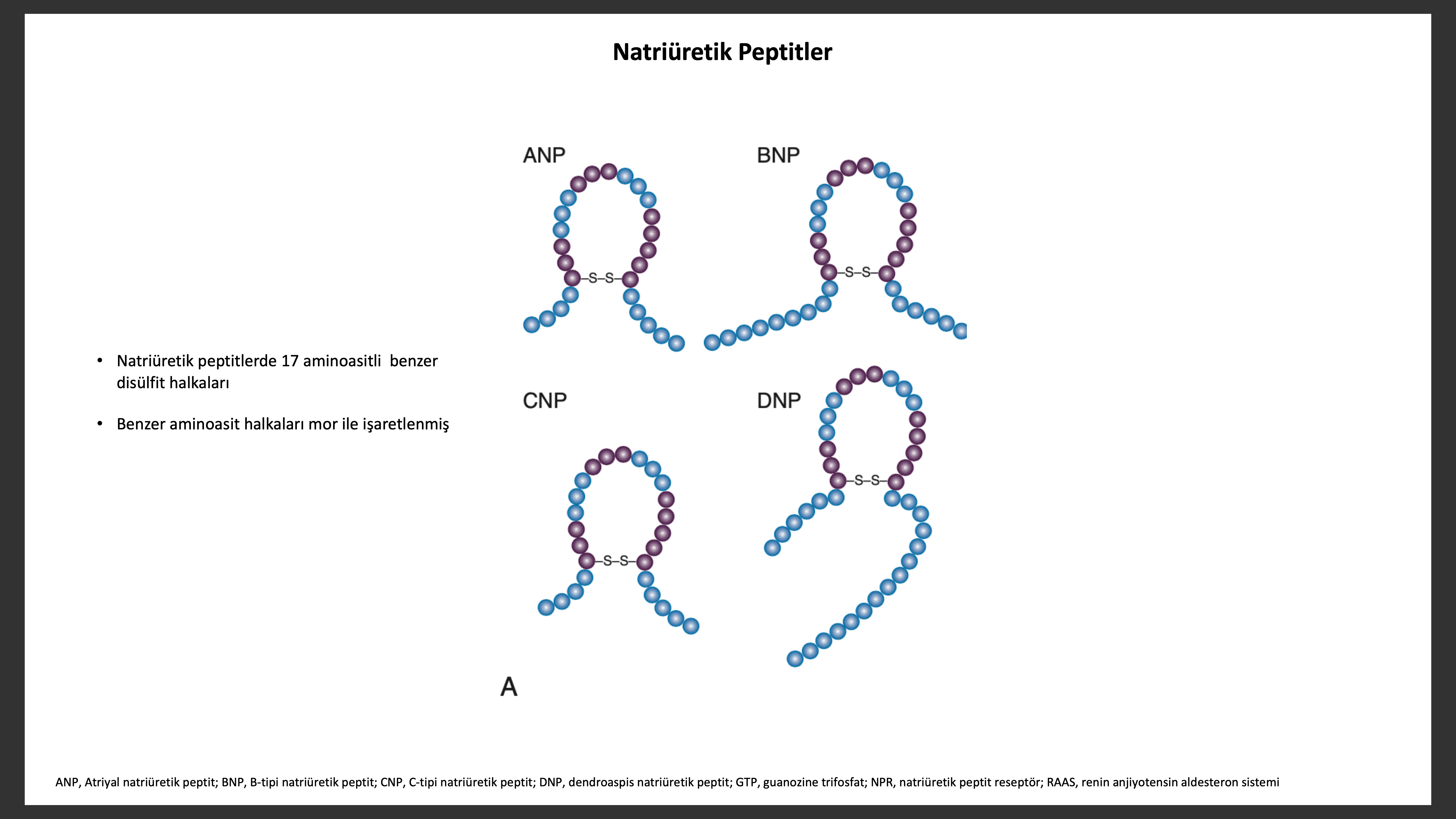
Natriüretik peptit sistemi yapısal olarak beş benzer peptit içerir; ANP, ürodilantin (ANP izoformu), BNP, C-tipi natriüretik peptit (CNP) ve dendroaspis natriüretik peptit (DNP). ANP, 28-aminoasit peptit hormonu prensip olarak kardiyak atriyada, orijinal olarak domuz beyninde izole edilmiş olan 32-aminoasitli BNP ise birincil olarak kardiyak ventriküllerde yapılır. ANP ve BNP nin her ikiside kalp duvar gerilimi artınca salınır ancak nörohormonlar (ör anjiyotensin II, ET-1, katekolaminler) veya fizyolojik faktörler (ör, yaş, cinsiyet, renal fonksiyon) salınımında rol alabilir. BNP nin biyosentez, sekresyon ve temizlenmesinin ANP den farklılıklar göstermesi bu iki natriüretik peptitin farklı fizyolojik ve patolojik rolleri olabileceğini düşündürür. ANP atriyal basınçtakiakut değişikliklere bağlı olarak kısa patlamalar şeklinde salınırken BNP atriyal ve ventriküler basınçta kronik yükselmeye bağlı transkripsiyon regülasyonu ile aktive olur. ANP ve BNP başlangıçta pro hormon olarak sentez edilir, corin ve furin tarafından yıkılarak biyolojik inaktif büyük N-terminal fragmanlarına (NT-ANP ve NT-BNP) ve küçük biolojik aktif peptitlere (ANP,BNP) ayrılır. ANP plazma yarı ömrü 3 dakika iken BNP ninki 20 dakikadır. Birincil olarak dolaşımda bulunan CNP de yine prohormon olarak salınıp biyolojik inaktif formu (NT-CNP) ve 22-amino asitli biyolojik aktif forma (CNP) ayrılır.
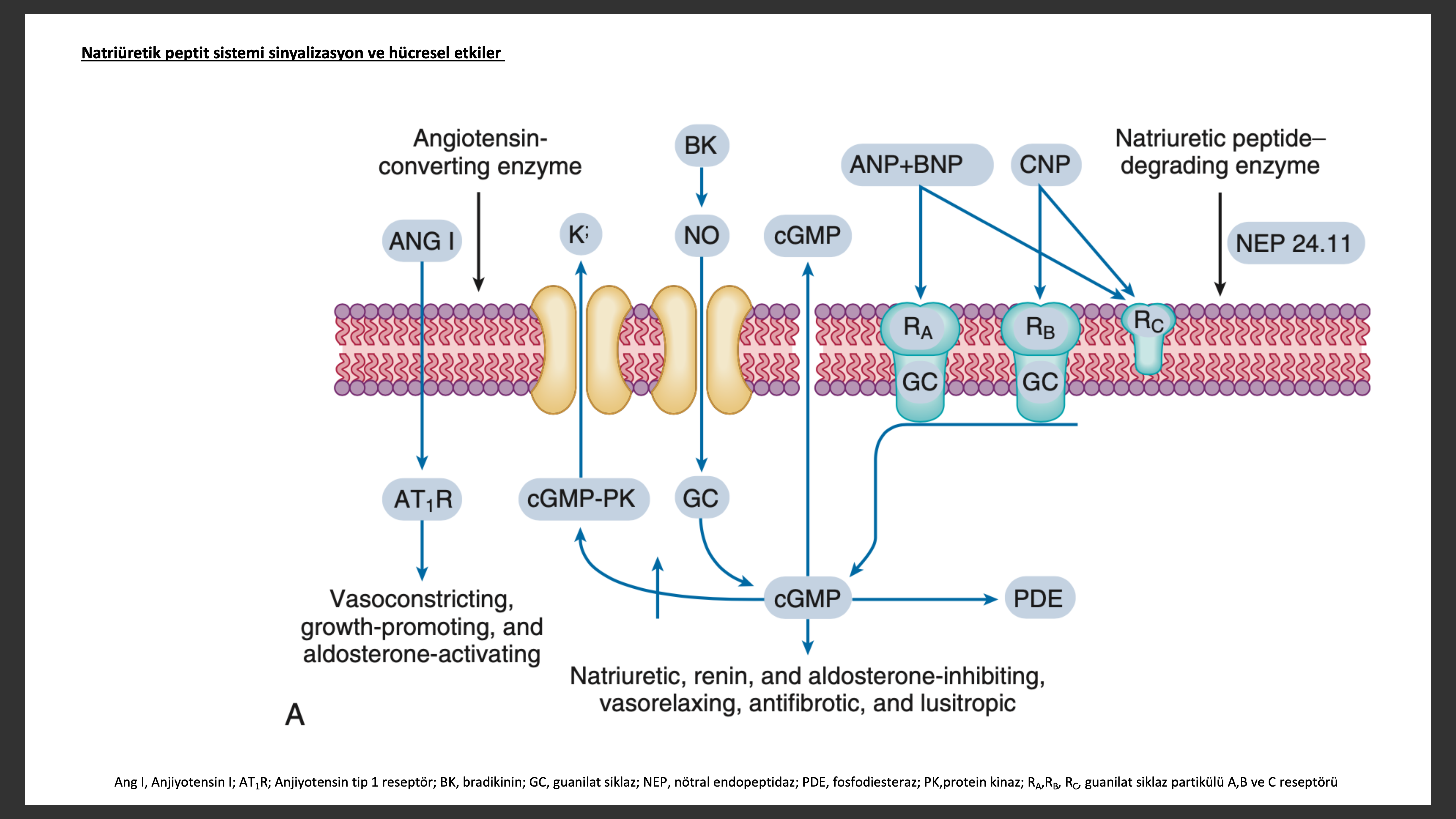
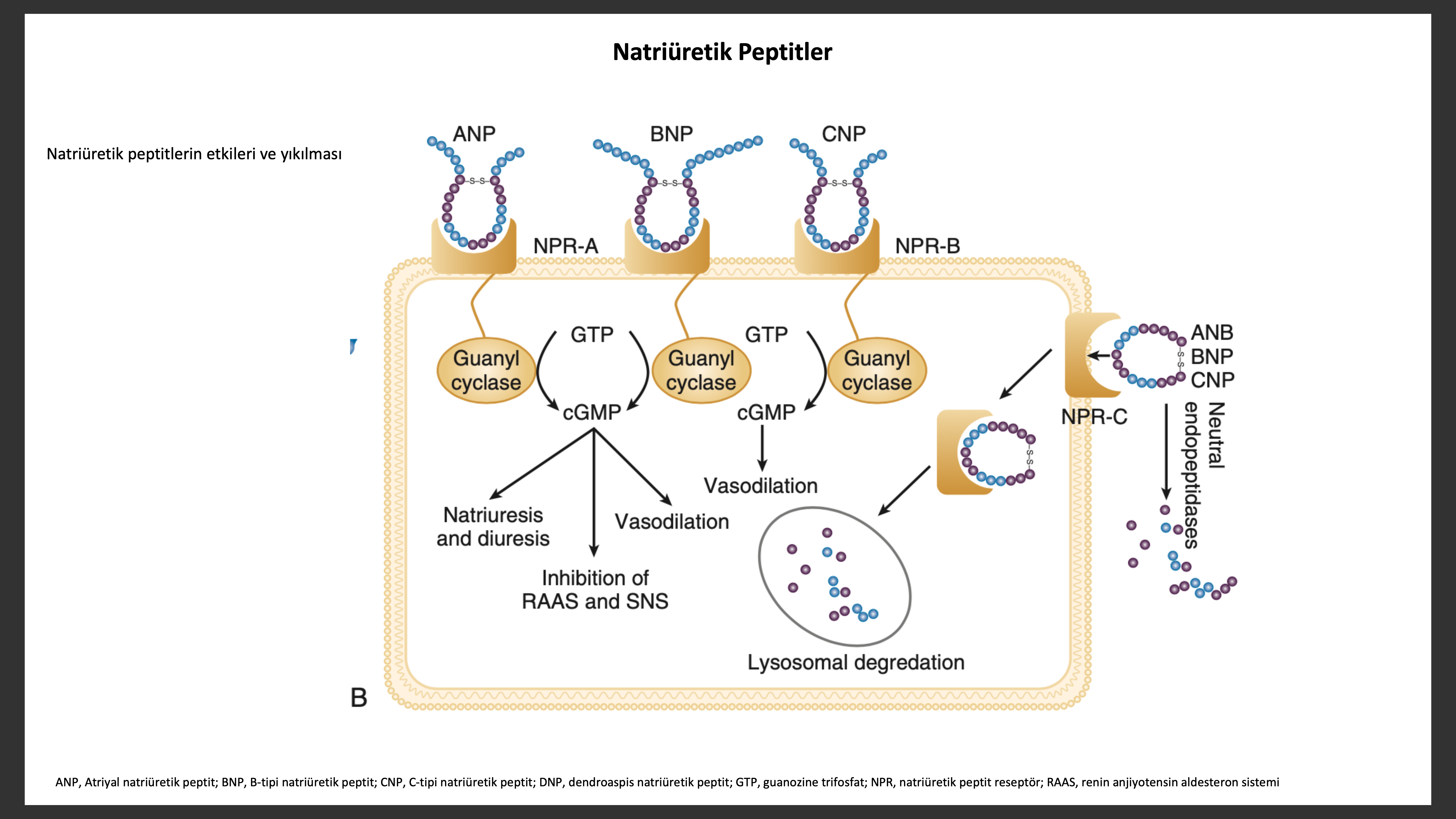 Natriüretik peptitlerin sinyal yolakları ise şu şekildedir. Natriüretik peptitler, natriüretik peptit A reseptörü (NPR-A) ile ANP ve BNP ye bağlanarak ve natriüretik peptit B reseptörü (NPR-A) ile CNP ye bağlanarak hücre içi ikincil ulak siklik guanozin monofosfatı (cGMP) aktive ederler. NPR-A ve NPR-B aktivasyonu natriürez, vazorelaksasyon, renin ve aldesteron ile fibrozis inhibisyonu ve artmış lusitropiye sebep olur. Natriüretik peptit C reseptörü (NPR-C) cGMP ile bağlantılı değildir natriüretik peptitler için temizleme reseptörü görevini görür. Tüm natriüretik peptitler majör olarak iki mekanizma ile yıkılır: NPR-C ile içeri alım takiben lizozomal yıkım ve nötral endopeptidaz (NEP) 24 ile enzimatik yıkım.
Natriüretik peptitlerin sinyal yolakları ise şu şekildedir. Natriüretik peptitler, natriüretik peptit A reseptörü (NPR-A) ile ANP ve BNP ye bağlanarak ve natriüretik peptit B reseptörü (NPR-A) ile CNP ye bağlanarak hücre içi ikincil ulak siklik guanozin monofosfatı (cGMP) aktive ederler. NPR-A ve NPR-B aktivasyonu natriürez, vazorelaksasyon, renin ve aldesteron ile fibrozis inhibisyonu ve artmış lusitropiye sebep olur. Natriüretik peptit C reseptörü (NPR-C) cGMP ile bağlantılı değildir natriüretik peptitler için temizleme reseptörü görevini görür. Tüm natriüretik peptitler majör olarak iki mekanizma ile yıkılır: NPR-C ile içeri alım takiben lizozomal yıkım ve nötral endopeptidaz (NEP) 24 ile enzimatik yıkım.
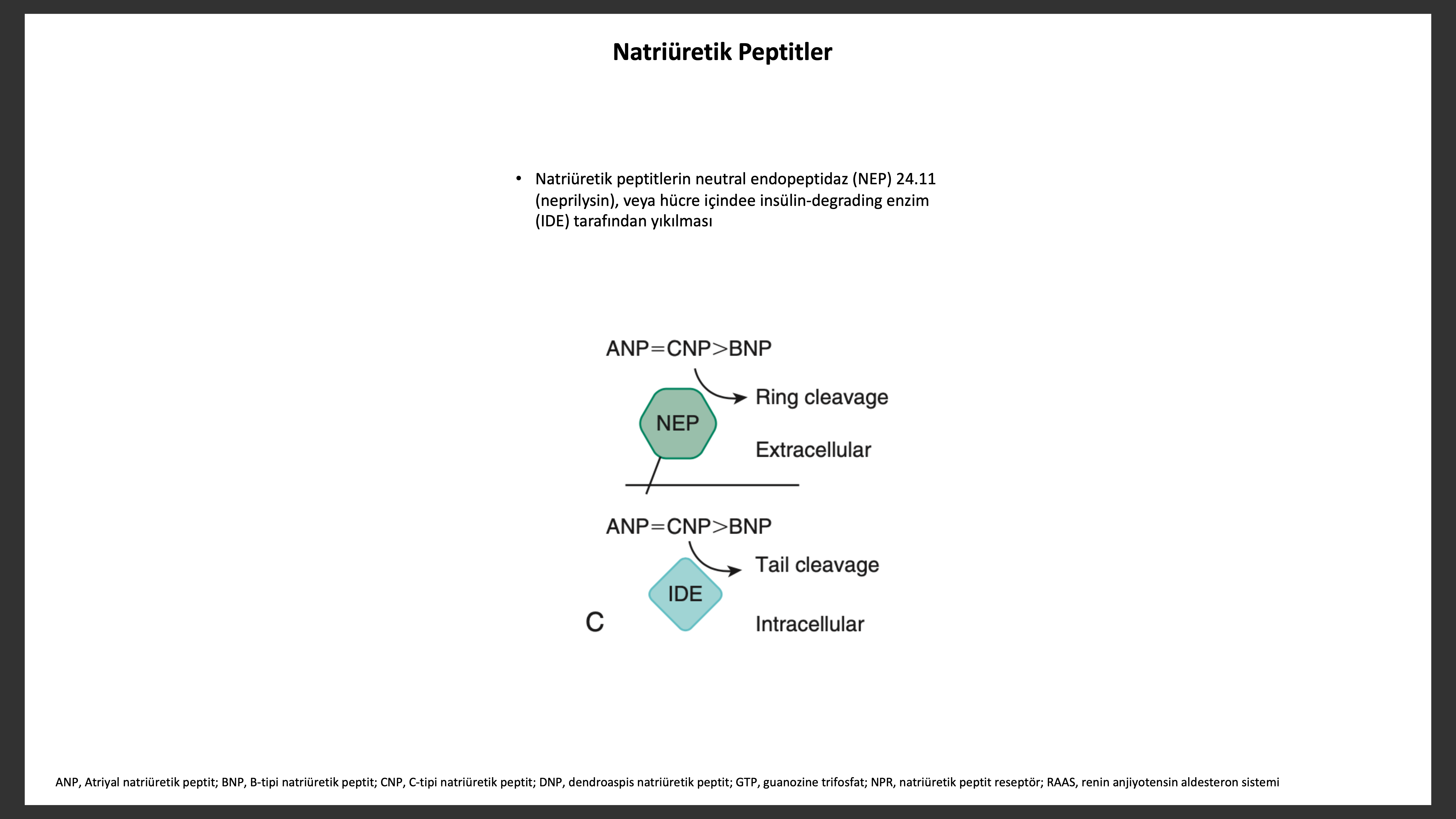 Neprisilin ACE ile yakın lokalizasyonlarda birçok dokuda bulunur. ACE ve NEP birçok biyolojik aktif peptit metabolizması ile ilişkili membran bağımlı çinko içeren metalopeptidazlardır. NEP tercihen hidrofilik rezidülerin N-terminalini ufak peptitleri böler. Vasküler endotel, düz adele hücreleri, miyositler, fibroblastlar, böbrek tübülleri ve sinir hücreleri gibi pek çok dokuda bulunur.
Neprisilin ACE ile yakın lokalizasyonlarda birçok dokuda bulunur. ACE ve NEP birçok biyolojik aktif peptit metabolizması ile ilişkili membran bağımlı çinko içeren metalopeptidazlardır. NEP tercihen hidrofilik rezidülerin N-terminalini ufak peptitleri böler. Vasküler endotel, düz adele hücreleri, miyositler, fibroblastlar, böbrek tübülleri ve sinir hücreleri gibi pek çok dokuda bulunur.
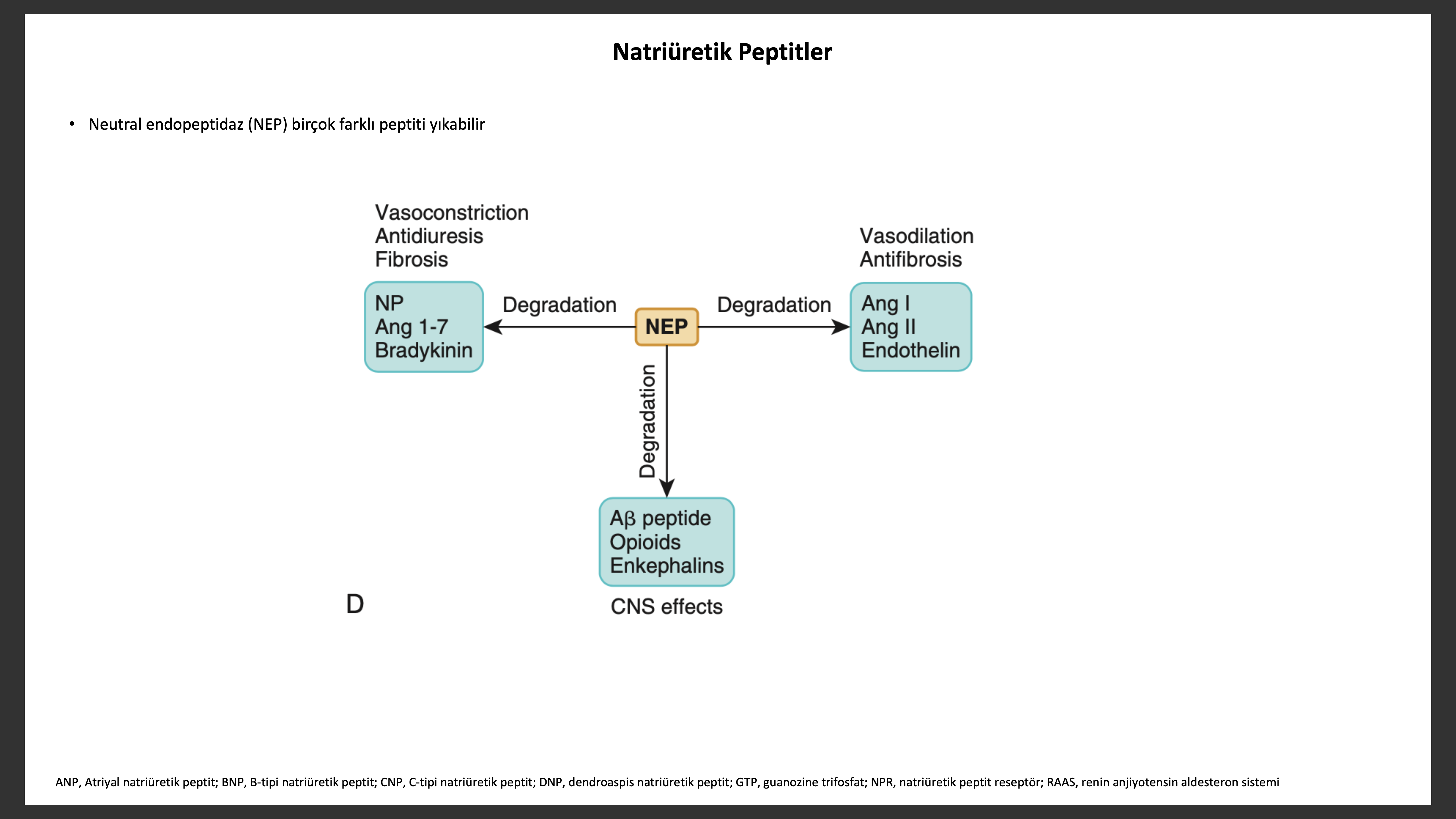
ACE ve NEP birçok biyolojik aktif peptit metabolizması ile ilişkili membran bağımlı çinko içeren metalopeptidazlardır. NEP tercihen hidrofilik rezidülerin N-terminalini ufak peptitleri böler. Vasküler endotel, düz adele hücreleri, miyositler, fibroblastlar, böbrek tübülleri ve sinir hücreleri gibi pek çok dokuda bulunur. NEP, natriüretik peptitler, anjiyotensin II, ET-I, adrenomedullin, opioidler, bradikinin, kemotaktik peptitler, enkefalinler ve amyloid-b peptit (Ab) gibi pek çok peptiti yıkar. NEP inhibisyonu ile natriüretik peptit yıkımının önlenmesi vazorelaksasyon, natriüreze, hipertrofi ve fibrozisin inhibisyonuna sebep olur. Diğer taraftan anjiyotensin II, anjiyotensin 1-7, ve ET yıkımının inhibisyonu natriüretik peptit vazodilatör etkisine ters olarak çalışır. Buna göre NEP inhibisyonu kan basıncı üzerine farklı etkiler yapacaktır.NEP inhibisyonu üriner kinin seviyelerini artırarak natriüretik etkiye katkıda buluna bilir. NEP beyinde amiloid peptit temizlenmesinde rol oynar. Özellikle NEP, nörotoksite ve Ab parçaları ile diğer proteinlerin birleşmesinden oluşan ve Alzheimer hastalığının izi olan amyloid plak oluşturan Ab yıkımında rol alır. Neprilysin fazlaca bulunması Alzhimer hastalığı gelişimini engellemiş, fare modellerinde kognitif fonksiyonu artırmıştır.
Kalp yetersizliğinde natriüretik peptitlerin potansiyel olumlu etkilerinden dolayı NEP inhibisyonunun kalp yetersizliği tedavisinde etkili olacağı düşünülmüş. Omapatrilat denilen ACE ve NEP dual vazopeptitaz inhibitörünün kalp yetersizliğinde ACE inhibisyonuna göre avantajı gösterilememiştir. Ancak AT1 reseptör antagonisti ve bir neprilysin inhibitörü kombinasyonu olan (valsartan/sacubitril, LCZ696) PARADIGM-HF çalışmasında hayat kalitesi, egzersiz kapasitesi, kalp yetersizliği sebebi ile hastaneye yatış ve total mortalitede belirgin azalma sağlamıştır. Natriüretik peptitlerin renal sodiyum ayarlamasındaki biyolojik önemi NPR antagonisti kullanan pek çok çalışmada gösterilmiştir. Deneysel kalp yetersizliği modellerinde NPR-A ve NPR-B akut blokajı veya kronik genetik NPR-A engellenmesi akut volüm artışına natriüretik yanıtı bozarak bize natriüretik peptit aktivasyonunun renal koruyucu etkisini göstermektedir. İnsanda kalp yetersizliğinde rekombinant insan ANP ve BNP infizyonu arteriyel ve venöz basıncı azaltarak kardiyak debi artışı ve nörohumoral aktivasyonun baskılanması gibi hemodinamik olarak olumlu etkiler göstermiştir. Ayrıca ANP ve BNP önemli tanısal ve prognostik bilgi vermektedir.
Nitrik Oksit
Serbest radikal gaz NO, NO sentaz (NOS) üç izoformu tarafından yapılır. Her üç izoform, NOS1 (nöronal NOS [nNOS]) dahil olmak üzere, NOS2 (uyarılabilir NOS [inducible, iNOS]) ve NOS3 (endotelyal-yapıdaki NOS [eNOS]). NOS1 kardiyak ileti sistemleri, kardiyak sinirler ve miyositlerdeki SR de bulunur. NOS normalde miyokartda bulunmaz ancak inflamatuar sitokinler varlığında kalpteki tüm hücrelerde yeni olarak yapılır. NOS3 koroner endotel, endokart, sarkolema ve kardiyak miyozit T-tübül membranlarında yapılır. NOS1 ve NOS3 kalsiyum veya kalmodilin ile uyarılırken, NOS2 kalsiyumdan bağımsızdır. NO çözünebilir guanilat siklazı aktive eder.
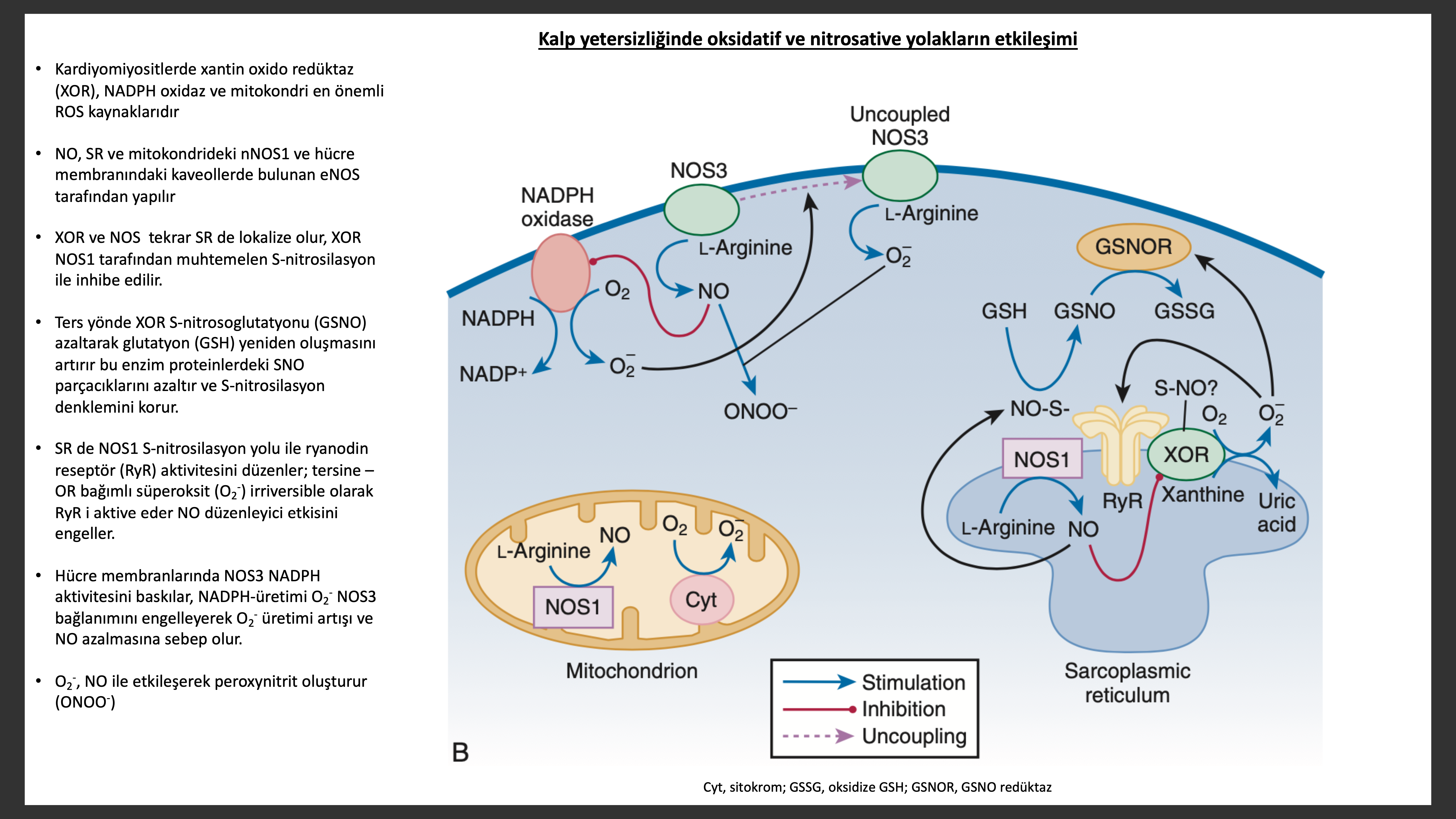
Normal durumda egzersiz sırasında endotelden sürekli salınan NO (endotelium-derive relaksasyon faktörü) vazokonstriktör etkilere ters vazodilatör yanıt yapar. Bu aktivasyon protein kinaz G (PKG) yi uyaran siklik guanizin monofosfat (cGMP) yapımını artırır ve farklı sinyal döngüleri çalışır. Normal kişilerde endotel hücrelerinden salınan NO periferik damarlarda vazodilatasyonu cGMP nin vasküler düz adelede üzerindeki vazodilatör etkisinden faydalanarak yapar. Kalp yetersizlikli hastalarda NO aracılı periferik vasküler dilatasyon körelir. Bu NOS3 miktarı ve aktivitesindeki azalmaya bağlanmıştır.
NO miyokart üzerine kısa dönemde fonksiyon ve enerji, uzun dönemde ise yapısal kompleks etkileri vardır. NO eksitasyon kontraksiyon eşleşmesinde ayrıca mitokondrial solunum kompleksinde rol alan kalsiyum kanalları üzerinde anahtar uyarıcı roller üstlenir. Bu düzenleme eksitasyon-kontraksiyon eşleşmesinde mikrolokalizasyonların NOS izoformları tarafından özel noktalardan etkilenmesi ile olur. Özellikle NOS1 ryanodin reseptörü (RyR) proksimal kısmı ile SR ve sarkoplazmik retikulum kalsiyum-adenozin trifosfat (SR Ca2+-ATPaz, SERCA2a) lokalizasyonunda ve NOS3 sarkolemmal kaveollerde hücre yüzey reseptörleri ile birlikte kompartman bölümlerinde ve L-tipi kanallarında bulunur. NO aynı zamanda eksitasyon-kontraksiyon enerji ihtiyacını karşılayan mitokondriyal solunumun işleminde rol alır. NOS ayrıca sol ventrikül yeniden yapılanmasında rol alır. NOS2 defekti olan transgenetik farelerde MI sonrası sol ventrikül yeniden yapılanmasının ve hayatta kalımın daha iyi olduğu görülür. Tam tersine MI sonrası NOS3 fazladan bulunması LV yeniden yapılanmasını olumlu etkiler. NOS2 ve NOS3 arasındaki farklı etkiler NOS2 nin NO salınımını miktar olarak çok daha fazla artırıyor olmasından kaynaklanıyor olabilir. Sol bulgular serbest radikal miktarı arttıkça NO miktarının azaldığını göstermekte , buna “nitroso-redox dengesizliği” denilmektedir. Tetrahidrobiyopterin defektine bağlı NOS bağlanma kusuru nitroso-redox dengesizliğine katkıda buluna bilir. Oksidatif stress ve NO periferal vazodilatör etkisini kaybolmasına bağlı nitroso-redox dengesizliği muhtemelen kalp yetersizliğinin ilerlemesine katkıda bulunur.
Bradikininler
Kininler, kallikreinin proteolitik enzim etkisi ile inaktif formdaki protein öncülü (kininojen)den dönüşerek salınan vazodilatörlerdir. Kininlerin biyolojik etkileri B1 ve B2 reseptörlerine bağlanarak ortaya çıkar. Çoğu kardiyovasküler etkisi B2 reseptörü üzerindendir, reseptör dokularda fazlaca bulunur ve braykinin ve kalidinine bağlanır. B1 reseptörü braykinin ve kalidinin metabolitlerine bağlanır. B2 reseptörü uyarılması NOS^, fosfolipaz A2 ve adenilylil siklazı uyararak vazodilatasyona sebep olur. Çalışmalar kalp yetersizliğinde vasküler tonüsün ayarlanmasında bradikininin önemli rol üstlendiğini göstermektedir. Bradikininin yıkımını artıran ACE ve neprisilin sadece potent vazokonstriktör (anjiyotensin II) yi artırmaz vazodilatör bradikinin salınımınıda inhibe eder. Bradikinin seviye artışı ACE ve NEP inhibitörlerinin etkisi ile olup kalp yetersizliğinde olumlu etki gösterir.
Adrenomedullin
Adrenomedullin orijinal olarak insan feokromistoma dokusunda keşfedilen 52-aminoasitli vazodilatör peptitdir. Takiben kardiyak atriyum, adrenal ve hipofiz bezlerinde yüksek, ventrikül, böbrek ve vasküler yapılarda düşük seviyelerde adrenomedullin immün reaktivitesi tespit edilmiştir. Adrenomedullin kalsitonin reseptör benzeri ve özgül adrenomedullin reseptörü dahil olmak üzere birkaç GPCRs ye bağlanır. Adrenomedullin reseptörleri endotelial ve vasküler düz adale hücrelerini de içeren pek çok doku yatağında bulunur. Kardiyak ve hemodinamik bozuklukla doğru orantılı olarak kardiyovasküler hastalıklar ve kalp yetersizliğinde adrenomedullin seviyeleri artar. Pek çok delil adrenomedullinin kalp yetersizliğinde aşırı periferal vazokonstriksiyon olumsuz etkisini ortadan kaldırarak kompanse edici etki gösterdiği yönünde. Kronik kalp yetersizliğinde plazma adrenomedullini yüksektir, seviyesi hastalığın ciddiyeti ile doğru orantılı olarak artar.
Biyomarkers in Acute Heart Failure (BACH) çalışmasında adrenomedullin prohormonu ölçen immünoassay yüksekliği kalp yetersizliğine bağlı ölümün öngörülmesi açısından anlamlı çıkmıştır.
Apelin
Apelin GPCR APJ için endojen ligant olan vazoaktif bir peptitir. APJ geni anjiyotensin AT1 reseptörüne çok benzer bir gen kodlar. Halbuki AJP anjiyotensin II ye bağlanmaz. Kardiyovasküler sistemde apelin endotel-bağımlı NO ile vazorelaksasyon ile arterial kan basıncını düşürür. Ek olarak apelin kardiyak miyosit hipertrofisine sebep olmaksızın kuvvetli inotropik aktivite gösterir. Ayrıca apelin arginin vazopresin aktivitesini inhibe ederek diüretik etki gösterir. Deney hayvanlarında yetmezlikli kalplerde apelin konsantrasyonları belirgin olarak düşük iken anjiyotensin reseptör blokör tedavisi sonrasında konsantrasyon belirgin artar. Apelin seviyeleri kalp yetersizliğinde kontrol guruplarına oranla belirgin düşüktür ve kardiyak resenkronizasyon tedavisi sonrası belirgin artar. APJ reseptörü çift fonksiyonlu bir GPCR dir, endojen ligantı uyarılınca kardiyoprotektör sinyal yollar ve mekanik sensör etkisi ile hemodinamik basınç yüklenmesi sonrası kardiyak hipertrofiyi sınırlar. CLR325 faz II klinik çalışmalar yapılan aperin reseptör agonistidir.
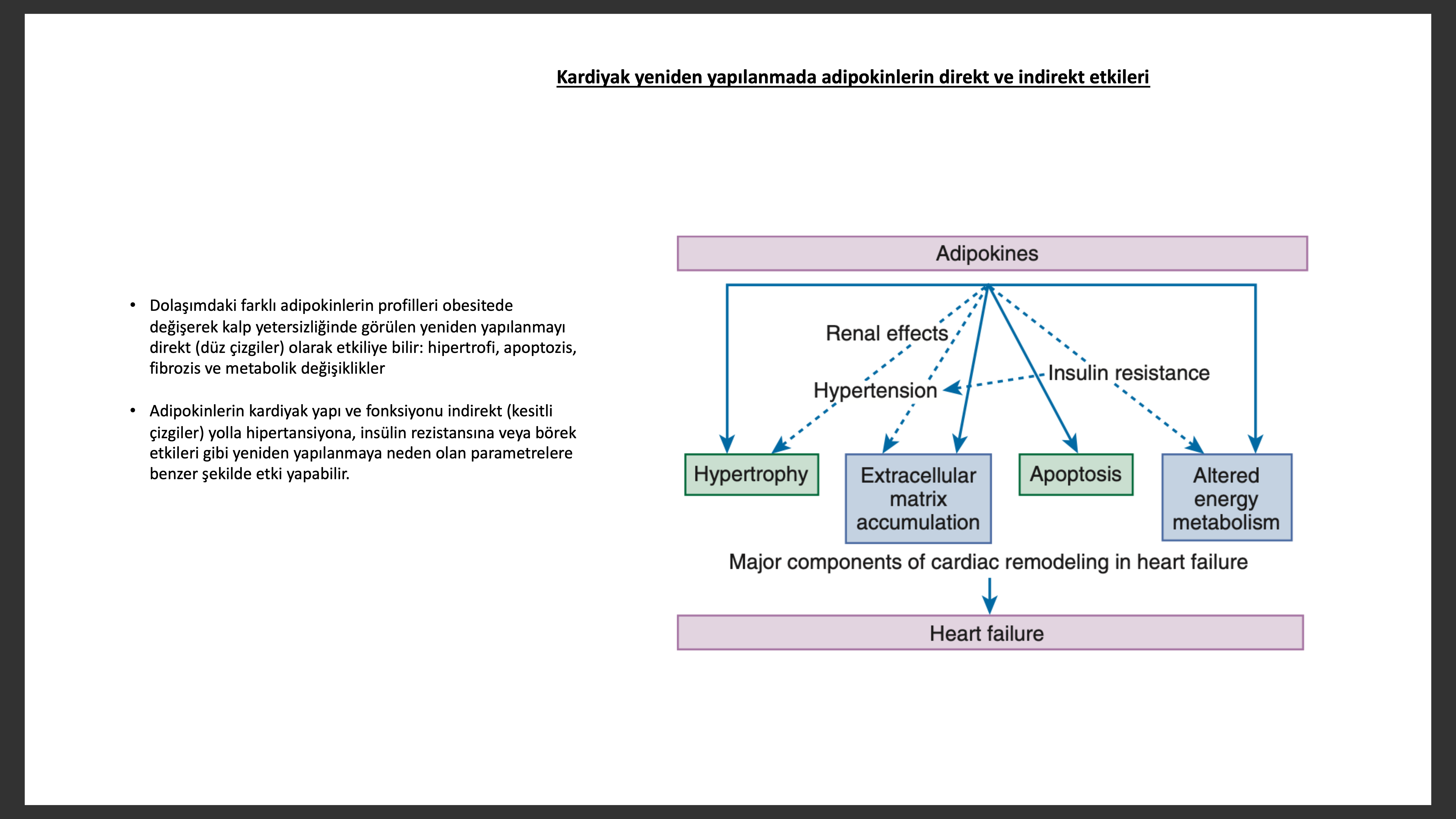
Adipokinler
Eskiden adipoz doku sadece basit bir yağ deposu olarak düşünülürken, artık adipokinler olarak topluca adlandırılan bir protein türünü sentez edip salgıladığı bilinmektedir. TNF, plazminojen aktivatör inhibitörü tip 1 (PAI-1), transorming growth faktör- b ve rezistin adipokinlerdir. 16-kDa lık bir protein hormon olan Leptin enerji alımı ve harcanmasında önemli bir düzenleyicidir. Ob geninin ürünü olan Leptin baskın olarak adipozitler tarafından yapılıp salınsa da kalpte de leptin sentez edilir. Leptinin öncelikli görevinin hipotalamik uyarı ile iştahı baskılamak ve besin alınımını azaltmak olduğu sanılırdı. Ancak dolaşımda artmış bulunan ve etkisini bir takım reseptör türleri (ob.R) üzerinden yürüten leptinin hipertansiyonda, hipertrofi ve kalp yetersizliğinde rolü olduğu görülmüştür. Leptinin miyokardiyal fonkssiyon üzerine etkisi periferal veya santral sinir sisteminde ikincil etkileri sonucu olabilir. Leptin eksikliğinde ve direncinde lipitler adipoz olmayan periferik dokularda birike bilir ve kardiyak adale apoptozisi de dahil bir takım “lipotoksik” etkiler yapabilir. Bazı çalışmalar insan ve kobay kardiyak miyositinde leptinin direkt olarak hipertrofi yaptığını göstermiştir.
Adiponektin, glukoz regülasyonu ve yağ asit oksidasyonu gibi metabolik süreçleri ayarlayan 224-aminoasitli polipeptitdir. Önceleri adiponektinin sadece adipoz dokuda olduğu düşünülürken son çalışmalar kalpte adiponektin varlığını göstermiştir. Adiponektin defekti olan fare çalışmalarında hemodinamik basınç yüklemesi sonrasında ilerleyici kardiyak yeniden yapılanma görülürken, hem normal hem de gen defektli farede, MI sonrası reperfüzyon ile MI alanında, apoptozda ve TNF yapımında azalma saptanmıştır. Obesiteye bağlı kalp yetersizliğinde pek çok çalışmada adiponektin seviyesinde azalma saptanmıştır. Adiponektinin kalp yetersizliğinde biyomarker tedavisinde teropetik hedef olarak kullanılması önerilmiştir.
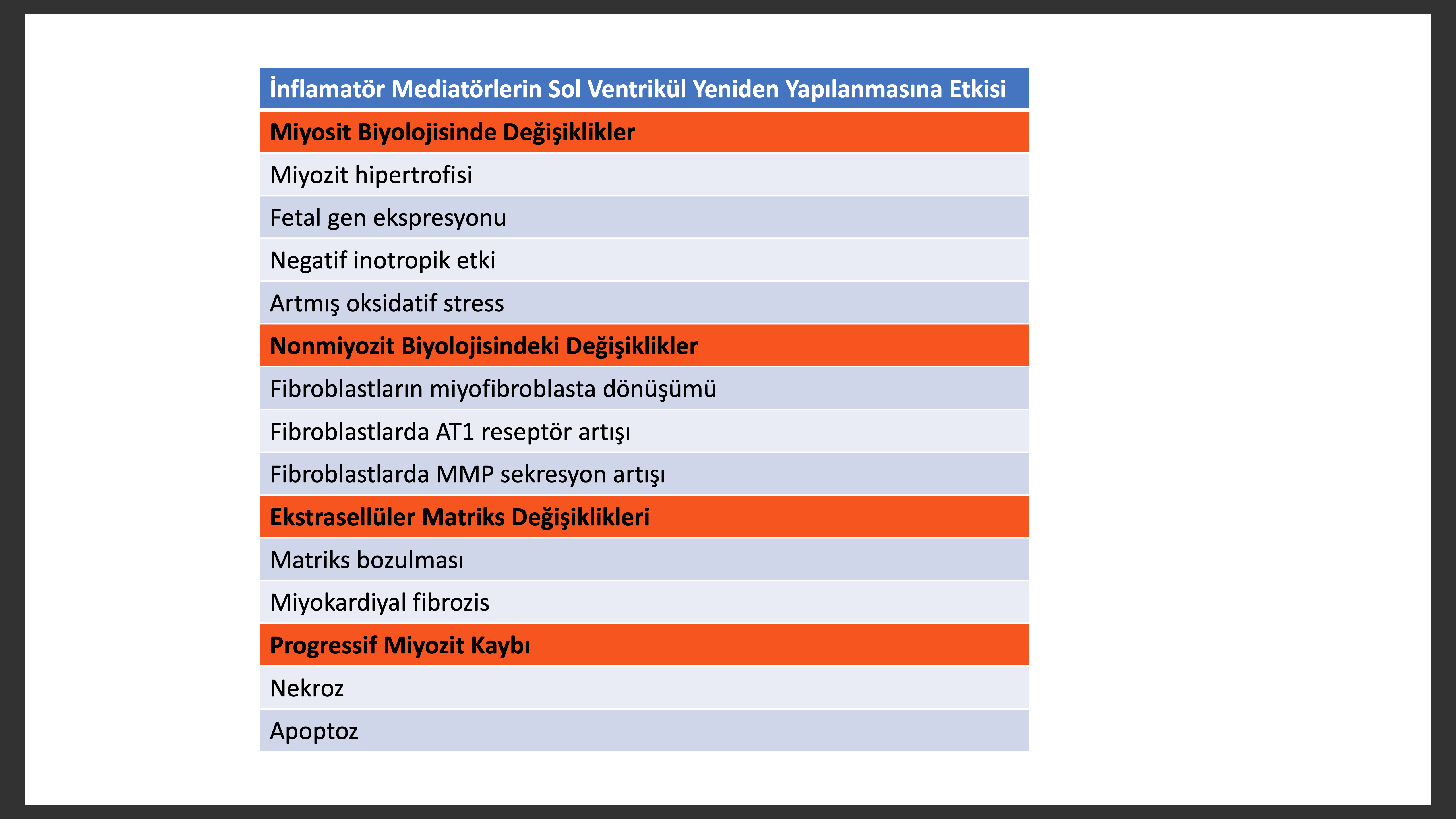
Inflamatuar Mediatörler
Yetişkin kalbi doku zedelenmesinde, hemostazı sağlayacak proteinlerin sentezini doku onarımı mekanizmalarını veya kalpteki kriptoprotektif mekanizmaları aktive ederek sağlar. Kalpte doku onarımını proinflamatuar doku topluluğunun doğal immün sistem üyeleri TNF, IL-1b ve IL-6 ,aşağı doğru akım “effektörleri” olarak etkiler. Bu miyokardiyal doğal immün yanıtının doku hasarı sonrası nasıl kordine olduğu yakın zamana kadar anlaşılamadı. Relatif olarak yeni keşfedilen Toll-like reseptör (TLRs) ve NOD-like reseptör (NLRs) ailesi, doğal immün cevabın yukarı doğru moleküler parçalarının nasıl düzenlendiğini anlamamızı sağladı. Bu moleküllerin birincil görevinin zedelenmiş miyokartın onarılması olmasına rağmen, uzun süre veya yüksek seviyelerde bu moleküllerin bulunması kalp yetersizliği fenotipini tüm yönüyle kardiyak miyosit, nonmiyosit ve ekstrasellüler matrikste olumsuz etkilerle yeniden şekillendire bilir. Ek olarak deneysel modellerde proinflamatuar sitokinler ile RAS arasında çapraz haberleşme vardır, örneğin anjiyotensin II nükleer faktör-kB (NF-kB)- bağımlı yolak vasıtası ile TNF baskınlığını artırır ve aynı şekilde inflamatuar mediatörlerin baskınlığı miyokardiyal ACE ve chymase yolu ile RAS ı artırırlar. Kalp yetersizlikli hastalarda proinflamatuar sitokinler (ör TNF, IL-6) artmıştır ve kötü gidişat habercisidir. Tersine anti inflamatuar sitokinler (ör; IL-10) kalp yetersizliğinde azalırlar, kalp yetersizliği ciddiyeti ve seviyesi arttıkça daha fazla azalır, bu durum pro ve anti inflamatuar sitokin baskınlığının hastalık seyrinde etkili olduğunu düşündürür.
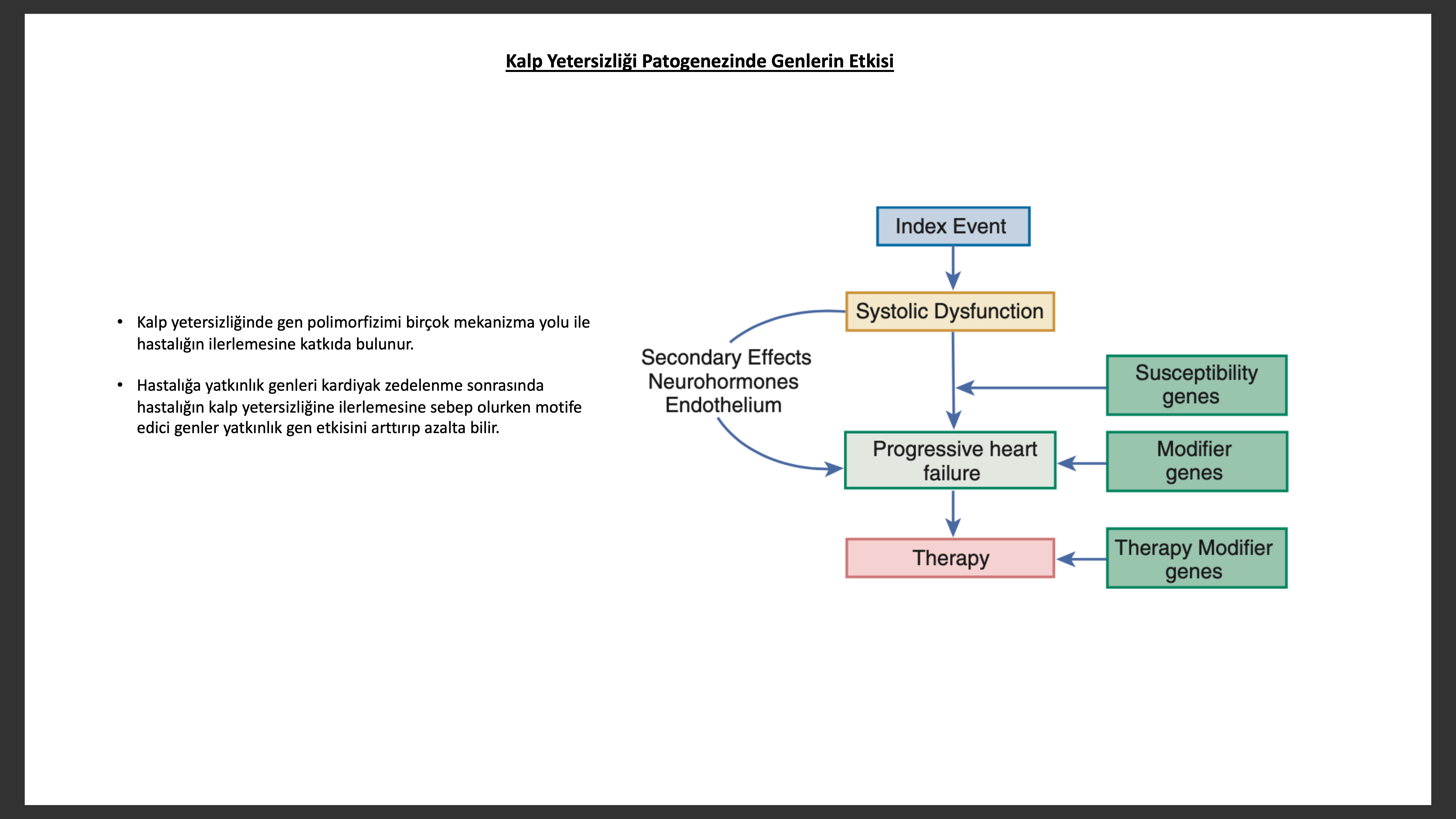
Sol Ventrikül Yeniden Yapılanması (Remodeling)
Nörohumaral model hastalık ilerlemesini büyük ölçüde açıklasa bile giderek artan klinik delil hazırdaki nörohumoral modelin hastalık ilerleme temelini tamamı ile açıklamadığı yönündedir. Her ne kadar nörohumoral sistemi inhibe eden antagonistler kalp yetersizliğinde hastalığın ilerlemesini engelleyip bir miktar geri çevirse bile çoğu hastada yavaş bir hızda olsa da hastalık ilerlemeye devam eder. Sol ventrikül yeniden yapılanması başlarda kalp yetersizliğinde olumlu gibi gözükse de ileri dönmede sol ventrikül performansını hastalığın klinik gidişini kötü yönde etkileyecektir (biyomekanik model). Sol ventrikül yeniden yapılanması hemodinamik, nörohumoral, epigenetik, genetik ve komorbit faktörlerden etkilenir.

Kalpte sol ventrikül yeniden yapılanması sırasında olan değişiklikler geleneksel tanımda anatomik olarak yapılsa bile yeniden yapılanma süreci kardiyak miyozitin biyolojisinde, miyozit ve miyozit dışı yapıların volüm ünde, sol ventrikül geometri ve mimarisinde değişiklikler yapar.
Kardiyak Miyozit Biyolojik Değişiklikleri
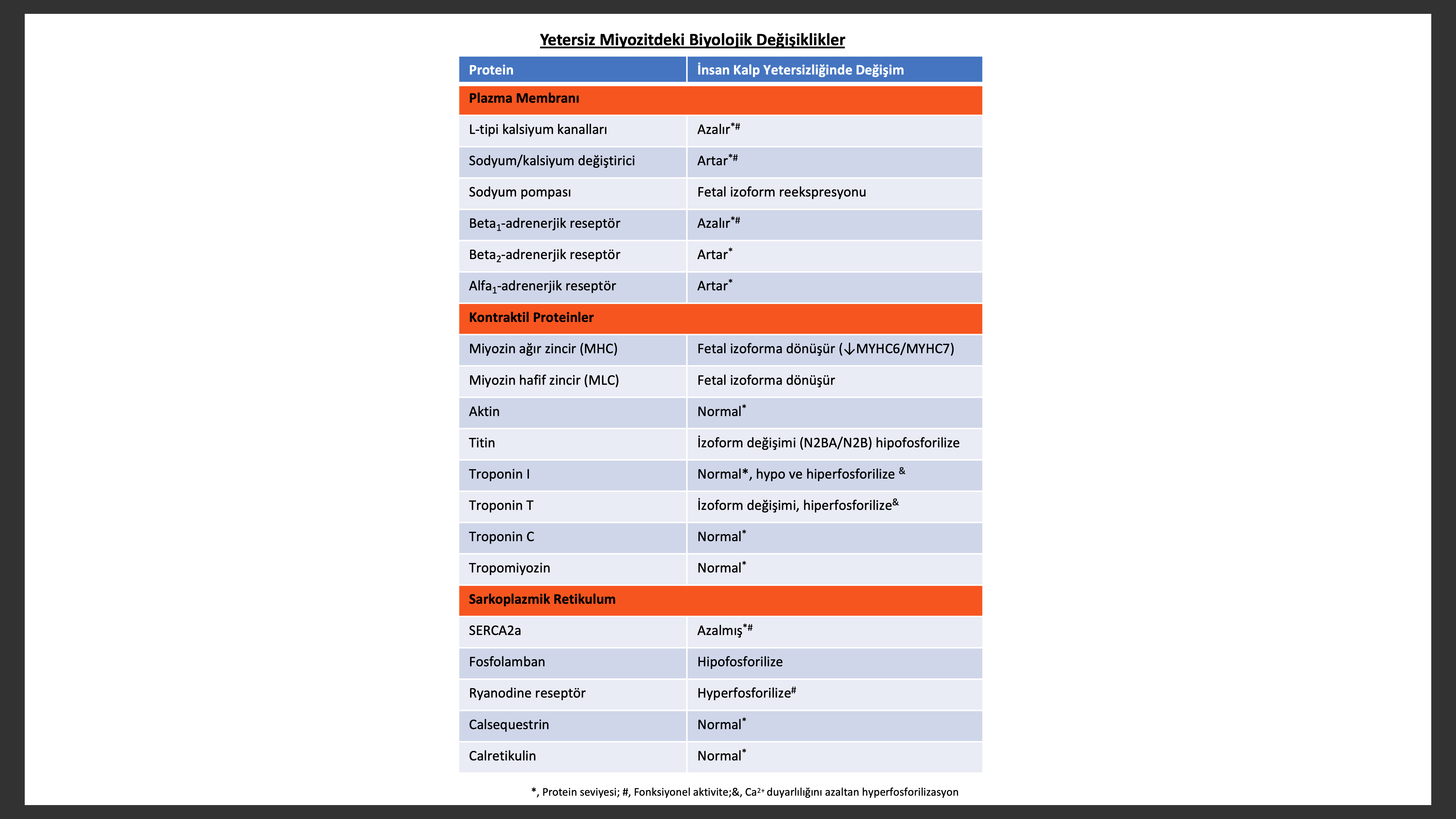
Çalışmalarda kusurlu kardiyak miyozitlerin önemli bazı değişikliklere uğrayarak kontraktil fonksiyonda ilerleyici kayıp olacağı öngörülmüştür. Bu değişiklikler alfa-miyozin ağır zincir gen ekspirasyon azalıp beta-miyozin ağır zincir ekspresyonunun artması, kardiyak miyozit miyoflamanlarında ilerleyen kayıp, cytoskeletal protein değişimi, eksitasyon kontraksiyon eşleşmesi ve enerji metabolizma değişiklikleri ve beta adrenerjik sinyallerde desensitizasyon şeklinde sıralana bilir.
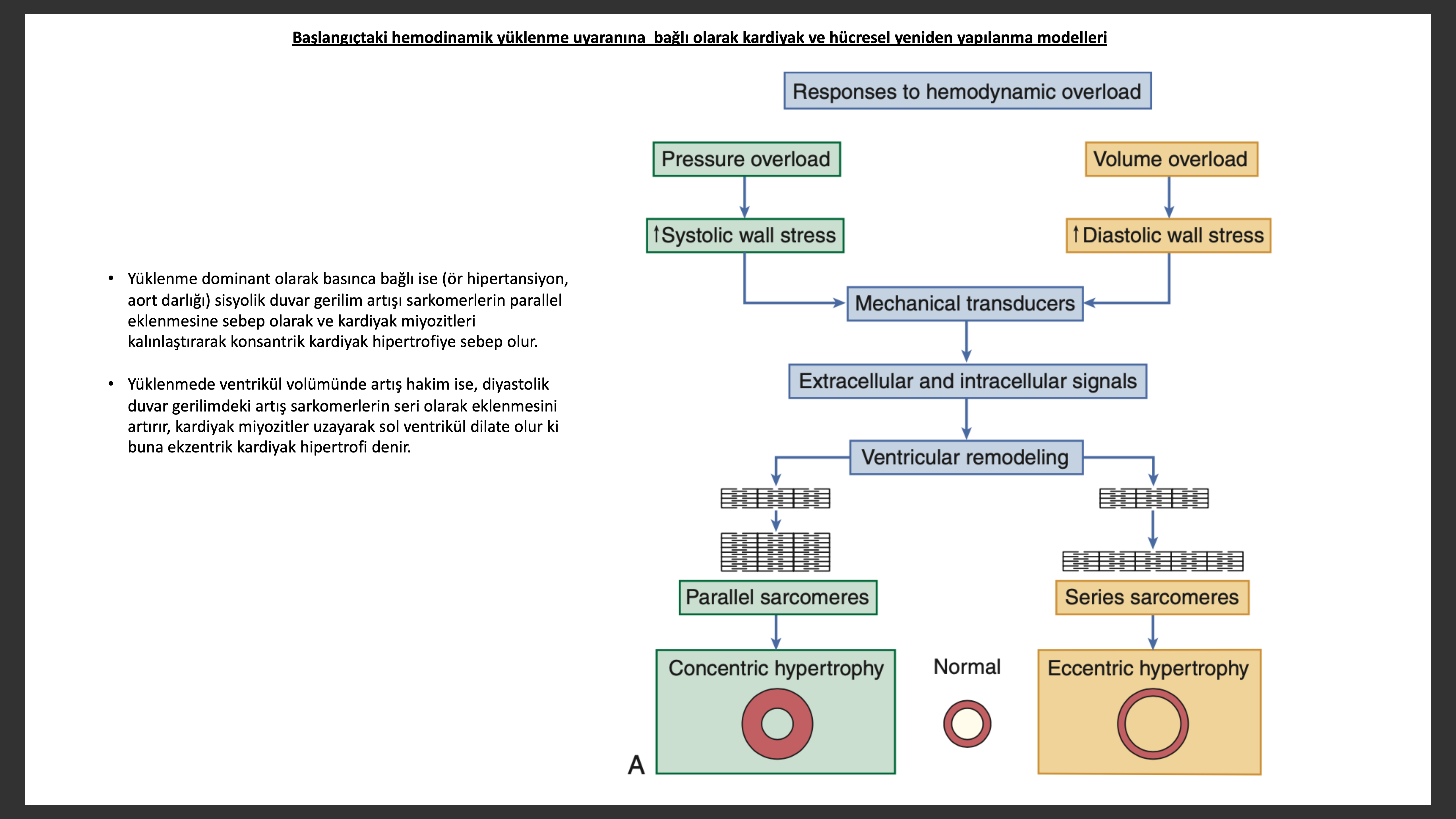
Kardiyak Miyozit Hipertrofisi
Hemodinamik yüklenmeye bağlı olarak iki temel hipertrofi olur. Basınç yüklenmesi hipertrofisinde (ör; aort darlığı veya hipertansiyon) artmış sistolik duvar direnci sarkomerlerin paralel eklenmesine sebep olarak, miyozitin kesitsel alanını artırarak sol ventrikül kalınlığını artırır. Bu şekildeki yeniden yapılanmaya “konsantrik hipertrofi” denilir ve Ca2+/kalmodulin protein kinaz II bağımlı sinyalizasyon farklılıklarından kaynaklandığı düşünülür. Tersine volüme yüklenme hipertrofisinde (ör; aortik ve mitral yetersizlik) diyastolik duvar gerilim artışı sarkomerlerin seri olarak eklenerek miyozit uzunluğunda artışa ve sol ventrikülde dilatasyona sebep olur. Bu şekilde olan yeniden yapılanmaya kalbin göğüs kafesindeki pozisyonuna bağlı olarak “egzentrik hipertfofi” denir ve protein kinaz B (Akt) aktivasyonu ile ilişkili bulunmuştur. Kalp yetersizlikli hastalar klasik olarak duvar kalınlığı azalmış veya normal dilate sol ventrikül içerirler. Bu görünümdeki yetersizlik olan kalpte kronik sıvı yüklenmesine bağlı miyositler karakteristik olarak uzamıştır.
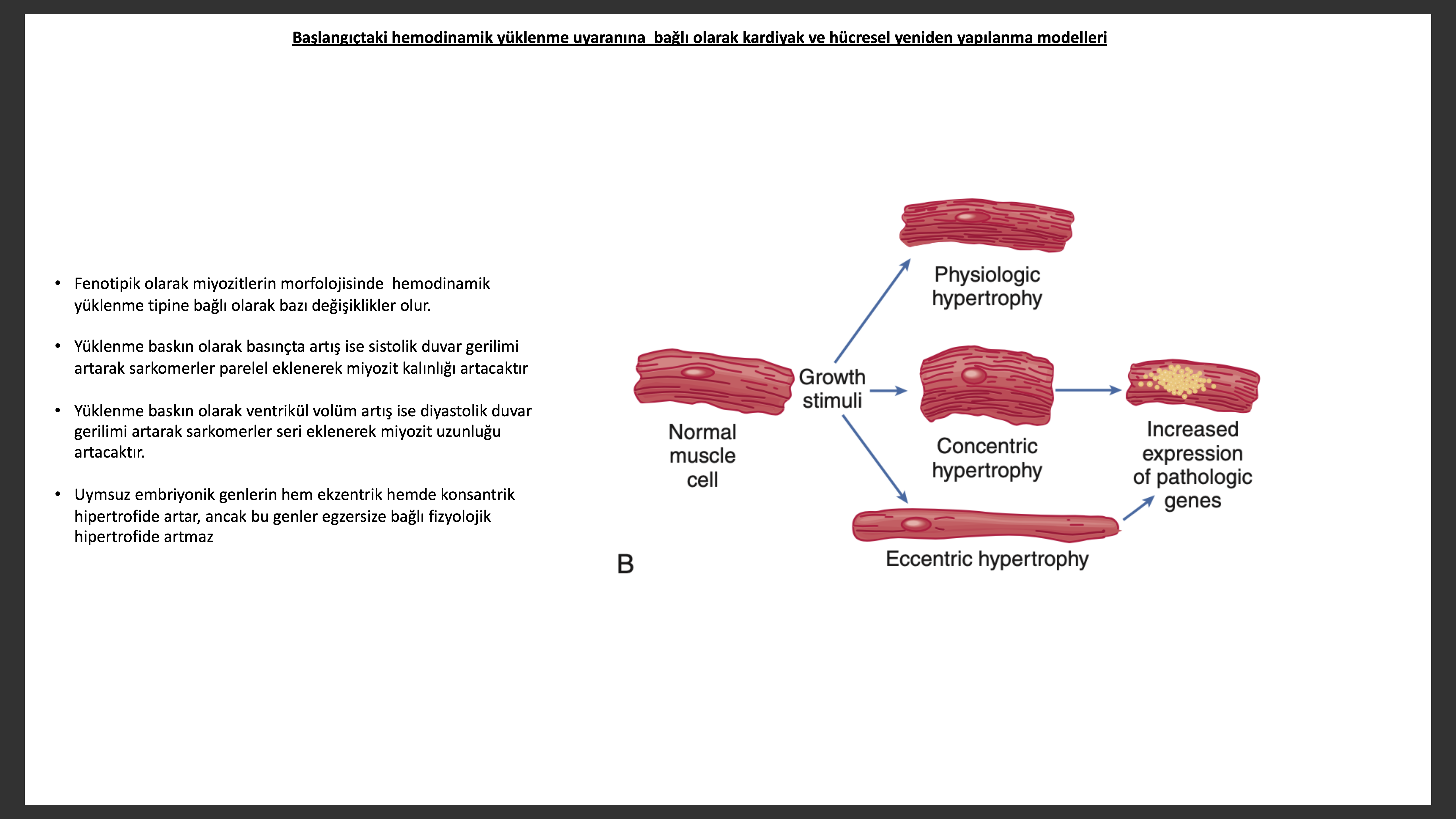
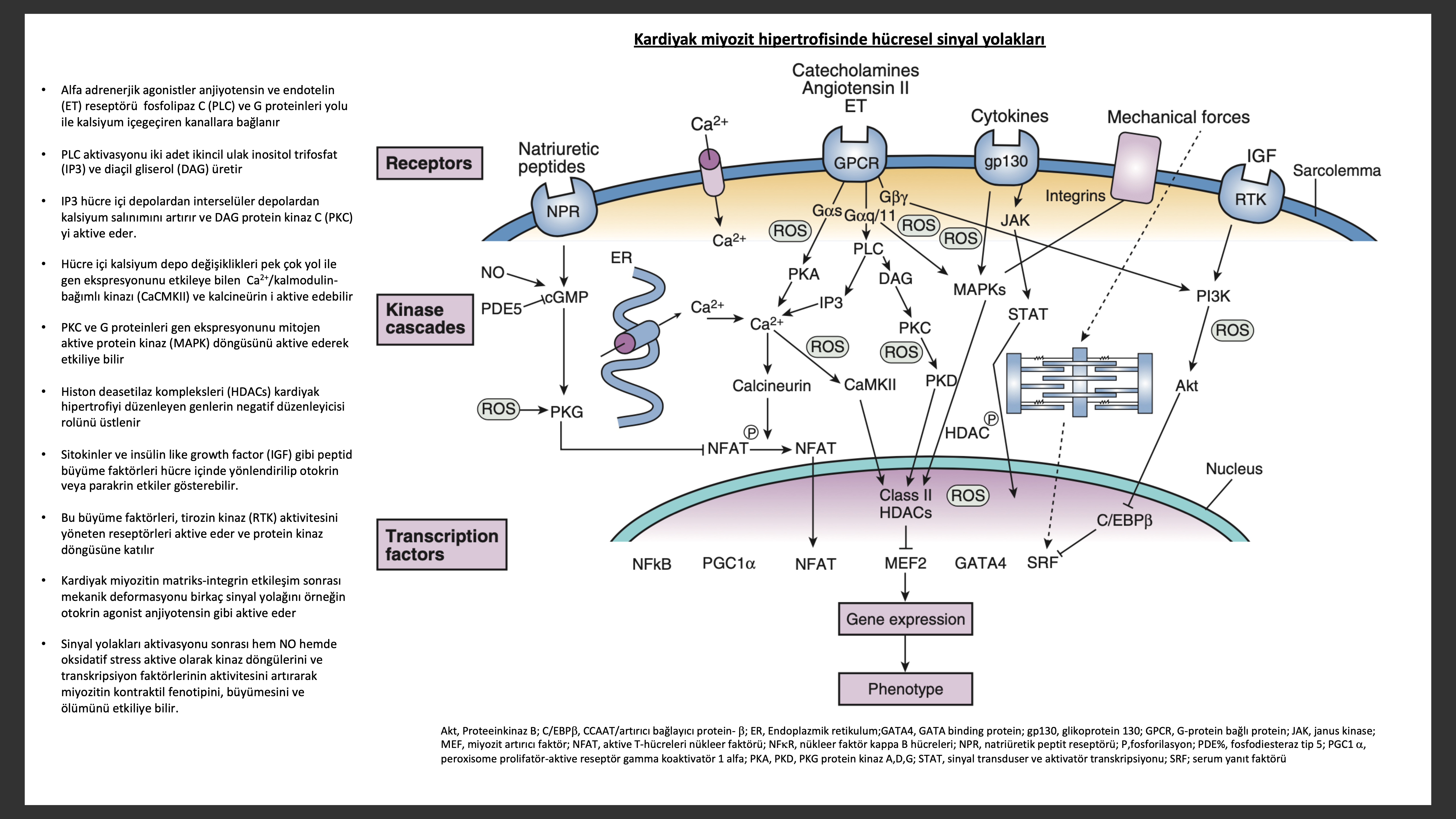
Kardiyak miyozit hipertrofisi biyolojik fenotipi değiştirerek normal olarak ekspresyonu olmayan postnatal gen portföylerinin reaktivasyonunu sağlar. Fetal gen programıda denilen bu fetal genlerin reaktivasyonu normalde yetişkin kalpte bulunan bazı genlerin ekspresyonunun azalmasına sebep olur. Sonra da bahsedileceği gibi fetal gen programı aktivasyonu yetersiz miyozite bağlı kontraktil fonksiyon bozukluğuna katkıda buluna bilir. Miyozitin genetik olarak tekrar programlanmasına miyozitdeki mekanik uzama/gerilme, nörohormonlar (ör; NE, anjiyotensin II), inflamatuar sitokinler (ör, TNF, IL-6), diğer peptitler, büyüme faktörleri (ör, ET) ve ROS (ör, süperoksit, NO) sebep olabilir. Bu etki hem miyokartda lokal otokrin/parakrin etki ile hem de sistematik endokrin etki ile olabilir. Kardiyak miyozit hipertrofisinde erken dönemde morfolojik olarak miyofibril ve mitokondride sayı olarak artış, ayrıca mitokondri ve nükleusda genişleme görülür. Bu evrede kardiyak miyozitler normalden büyüktür ancak hücre içi organizasyon korunur. Hipertrofi devam ettikçe mitokondri artacak ve yeni kontraktil elemanlar hücrede lokalize yerlere eklenecektir. Uzun süreli hipertrofiye uğrayan hücrelerde fazlaca lobüllü membranlı ileri derecede büyük nükleus, yakınındaki miyofibril yerdeğiştirmesi ile normal Z-bant yapı bozulması hücresel organizasyonda belirgin bozulmalara sebep olur. Hipertrofinin daha ileri evrelerinde kontraktil eleman kaybı (miyositolizis) , Z-bantlarında ileri bozukluk, yine sarkomer normal parelel diziliminde bozukluk, eşlik edenT tübül dilatasyonu ve kıvrıntılı hal alması görülür.
Eksitasyon- Kontraksiyon Eşleşme Değişiklikleri
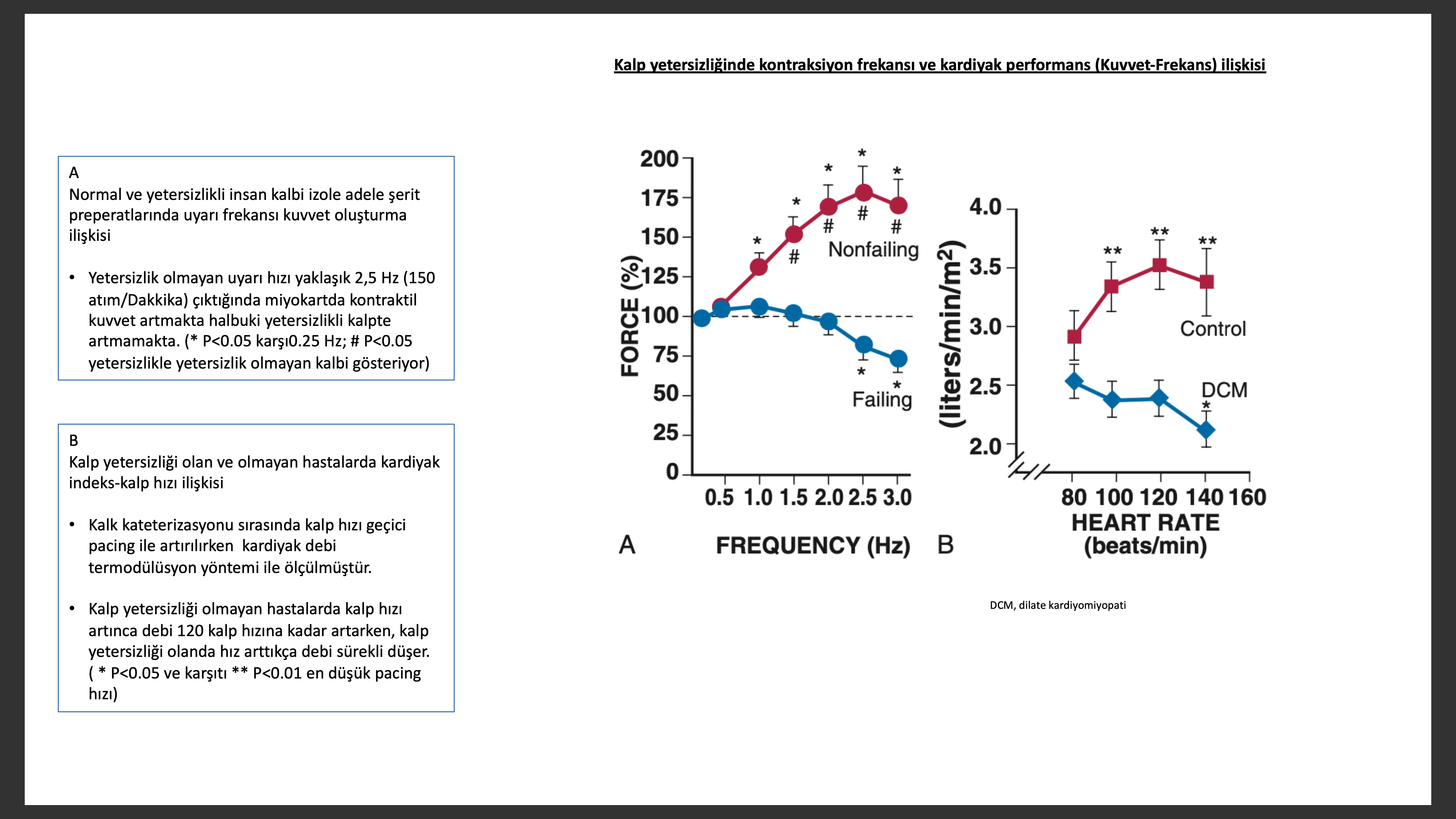
Eksitasyon kontraksiyon eşleşmesi kardiyak aksiyon potansiyeli ile başlayan ve miyozit kontraksiyon ve relaksasyonu ile biten biyolojik olayların döngüsüdür. Yetmezlik olan kalpte azalan kontraksiyon ve relaksasyon en çok yüksek kalp hızlarında belirgindir, azalmış kuvvet frekans ilişkisi görülür. Bu hem izole insan miyokart çalışmalarında ve klinik gözlemlerde gösterilmiştir. Normalde yüksek kasılma frekansı, frekans bağımlı hücre içi kalsiyum geçiş artırarak kardiyak performansı artırır. Aksine yetersizlik olan miyokartda yüksek kalp hızlarında hücre içinde Ca2+ azalması, uzamış Ca2+ geçiş azalması ve diyastolde hücre içi Ca2+ artması bağlı kuvvet üretiminde azalma olur. Hücre içinde Ca2+ geçişinde azalma , SR de Ca2+azalmasına ikincil olur. Bu kalsiyum döngüsünde olan 3 önemli faktöre bağlıdır.
Artmış ryanodine reseptörleri (RyRs) Ca2+ sızıntısı.
SERCA2a (SR kalsiyum pompası) protein seviye ve fonksiyon azalmasına bağlı SR Ca2+geri alımında azalma
Sarkollemal Na+/Ca2+ değiştirici (NCX) ekspresyon ve fonksiyonunda artma.
Artmış Ca2+ sızıntısı
Aksiyon potansiyel sırasında Ca2+hücre içerisine L-tipi kalsiyum kanalları ile girip, SR ve RyRs den daha fazla kalsiyum salınımına sebep olur. Her ne kadar kalp yetersizliğinde RyRs dışavurumu ve RyRs ile L tipi Ca2+kanal bağlantıları konusunda fikir birliği olmamasına rağmen, kalp yetersizliğinde Ca2+sızıntısının RyRs nin diyastolde açılmasından kaynaklandığına dair fikir birliği vardır. Sonuç olarak SR den kalsiyum salınımı Ca2+ kıvılcımı ismini alır. Kalp yetersizliğinde diyastolik Ca2+ sızıntısında altta yatan mekanizması , protein kinaz A(PKA), Ca2+/ calmodulin-bağımlı protein kinaz II (CaMKII) nin RyR fosforilizasyonunu artırması ve RyR nin sabitleyen protein calbastin (FKBP 12.6) bağlanmasını engellemesi. Çalışmalar PKA-bağımlı RyR nin fosfrorilasyonunun, calstabin ve FKB ilişkisini kararsız hale getirerek Ca2+sızıntısını artırdığını düşündürmektedir. İlginç olarak köpeklerde beta adrenerjik blokörler, RyR nin FKB12.6 ile ilişkisini tekrar kararlı hale getirerek Ca2+sızıntısını azaltmaktadır. Bu gözlem betablokör tedavisi sonrası kontraktil fonksiyonların düzelmesinin RyR kararlılığına bağlı olduğunu düşüdür. PKA-bağımlı RyR fosforilasyonunun artmasının kalp yetersizliği etiyolojisindeki önemi, kalp yetersizliğinde beta blokör reseptörleri azaldığı için paradoks gözükür. Önerilerden bir tanesi RyR ye yakın mikrolokasyonlarda PKA fosforilasyonu ve siklik adenozin monofosfatın (cAMP) artıp tip 4 fosfodiesteraz (PDE4D3)aktivitesinin azalmış olmasıın etkili olabileceği şeklindedir. SR Ca2+ içeriğinde azaltıcı etkisi dışında artmış sızıntı kalp yetersizliğindeki aritmiler içinde uygun zemin hazırlar. Bunun sebebi NCX aktivasyonudur: SR den sızan Ca2+ NCX i sitozelden Na+ karşılığında Ca2+ çıkarması için aktive eder. NCX elektrik yükleyici olduğu için (3 Na+ karşılığı 1 Ca2+), net içeri doğru akım geliştirir ki buna geç depolarizasyon (DADs) denir ve aritmileri tetikleyebilir. RyR ye bağlanıp stabilize edebilen diltiazem türevi JTV 519 gibi ilaçlar (RYCALS denir) deneysel kalp yetersizliğini ve aritmileri engellediği gösterilmiştir ve şu anda bir tedavi edici ilaç gurubu olarak ortaya çıkmıştır.
Sarkoplazmik Retikulum Ca2+ Geri Alımı ve Sarkolemmal Ca2+ Eliminasyonu
Kontraktil proteinlerin relaksasyonu Ca2+ troponin C ayrışması ve Ca2+sitozolden temizlenmesi sonrası başlar. İnsan kalbinde sitozolden Ca2+temizlenmesinde iki mekanizma rol alır. SR um SERCA2a Ca2+pompası ile kalsiyum geri alması ve NCX yolu ile transsarkolemmal Ca2+temizlenmesi. Normal şartlarda %75 Ca2+ SR ve % 25 NCX ile hücre dışarısına çıkarılır. Kalp yetersizliğinde SERCA2a protein seviye ve fonksiyonunda azalmaya bağlı SR Ca2+geri alımı azalır. Ek olarak yetersizlikte kalpte fosfolamban (PLB) fosforilasyonu azalır, SR Ca2+pompası PLB-bağlı inhibisyonu artar. SR Ca2+ geri alımının yetmezlikteki kalpte azalması NCX tarafındaneliminasyon azalması sebebi ile transsarcolemal Ca2+artar, bunun sebebi muhtemelen NCX protein ekspresyonunun artmasıdır.
Deneysel olarak gen transferi ile SERCA2a defektinin düzeltilmesi kontraktil fonksiyonu artırmış ve elektriksel kararlılığı sağlamıştır. Yeni CUBID çalışmasında gen transferi güvenli olsa bile SERCA2a transferi kalp yetersizliği hastalarda klinik fayda sağlamamıştır. NCX aktivitesindeki artış, miyozitden Ca2+ eliminasyonunu artırıp diyastolik Ca2+ seviyelerini koruyup diyastolik fonksiyon bozukluğunu engellese de SR Ca2+ alımı azalır ise NCX in artış aktivitesi, SR Ca2+ birikim/içeriğini daha da azaltarak kontraktil protein Ca2+aktivasyonunu azalta bilir. Daha öncede söylendiği gibi NCX elektrik yükü artırması DADs ve aritmilere sebep olur.
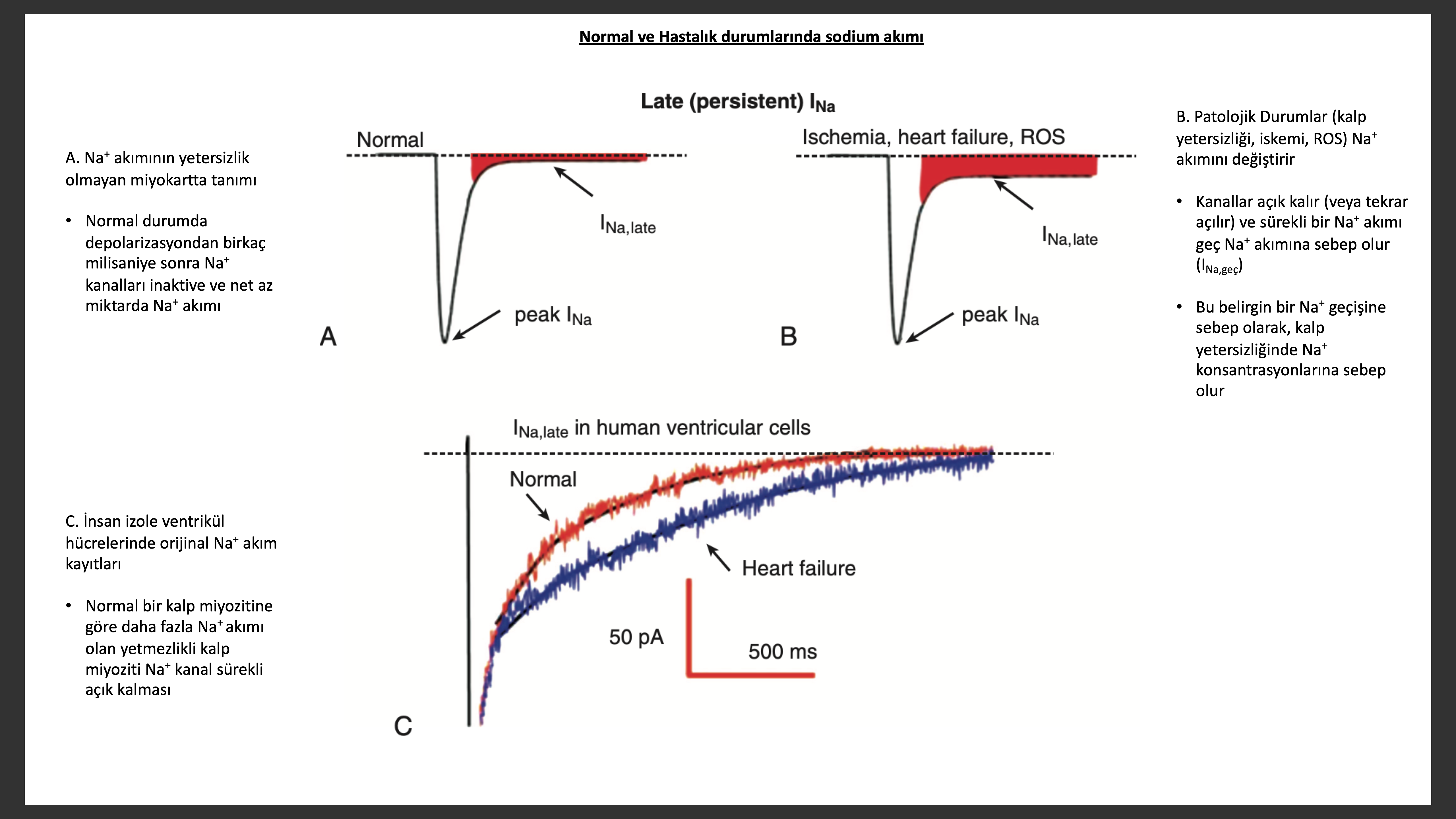
Aksiyon Potansiyel Süresince ve Sodyum yönetimi
Yetersiz kalpte yaygın olarak görülen artmış aksiyon potansiyel süresine bazı faktörler katkıda bulunur. Dışarı doğru geçici potasyum akımı (Ito) ve içeri doğru doğrultucu potasyum akımı (Ik1) kalp yetersizliğinde azalmıştır. Ek olarak içeri doğru NCX ile artmış Na+ akımı ve sodyum kanalının sürekli artmış aktivitesi aksiyon potansiyeli artmasına katkıda buluna bilir. Sonraki “gecikmiş sodyum akımı” denilen mekanizma kalp yetersizliğinde aritmilerin ortaya çıkmasına neden olabilir. Daha öncede belirtildiği üzere voltaj-kapı Na+ kanalları hücre membranı depolarizasyonu sırasında aktive olarak, hızlı içeri Na+ akımına sebep olur, bu aksiyon potansiyeli hızlı yukarı vurusundan (upstroke) sorumludur. Normal koşullarda Na+ kanalları depolarizasyondan birkaç milisaniye sonrasına kadar aktif değildir. Ancak artık biliniyorki bazı Na+ kanalları aksiyon potansiyeli plato evresinde açık kalmakta (veya yine açılır), ufak ama sürekli Na+ içeri akımı olup “geç” sodyum akımına (INa) sebep olmaktadır. Kalp yetersizliğinde INa azımsanmayacak miktarda sodyum hücre içine girişini artırarak aksiyon potansiyeli uzatır ve erken gecikmiş depolarizasyonlara sebep (EADs) olarak aritmiler için önemli bir kaynak olabilir. Hücre içi yüksek Na+ seviyeleri artmışı sodyum-proton değişim aktivitesine bağlı olarak hücrede asidoza sebep olabilir. Hücre içi Na+ artışı ayrıca NCX çalışma gücünü etkileyerek NCX ileri doğru etkisi ile Ca2+atılımını kalp yetersizliğinde azaltır buna SERCA2a pompa aktivite azalması da eklenir ise diyastolik sitozolik kalsiyum seviyeleri artırarak diyastolik fonksiyonu bozarlar. Yetmezlikli insan kalbinden izole edilmiş miyokartda geç Na+ kanallarını inhibe eden ranolizin bozulmuş diyastolik fonksiyonu düzeltirken antiaritmik etki de gösterebilmektedir. Hastadan hastaya Ca2+ yönetimi oldukça farklılık gösterebilir, kalp yetersizliği fenotipindeki farklılıklara muhtemelen bu sebep olur. Eğer SERCA2a ekspresyonu azalmış ve hücre içi sodyum yüksek ise hem sistolik hemde diyastolik fonksiyon bozulacaktır.Tersine NCX ekspresyonu orta derecede artar ise artmış olan Na+ transsarkolemmal kalsiyum eliminasyonunu artırar ve diyastolik fonksiyonlar korunur. Ancak NCX aktivitesine bağlı olarak bu sefer aritmiler arta bilir.
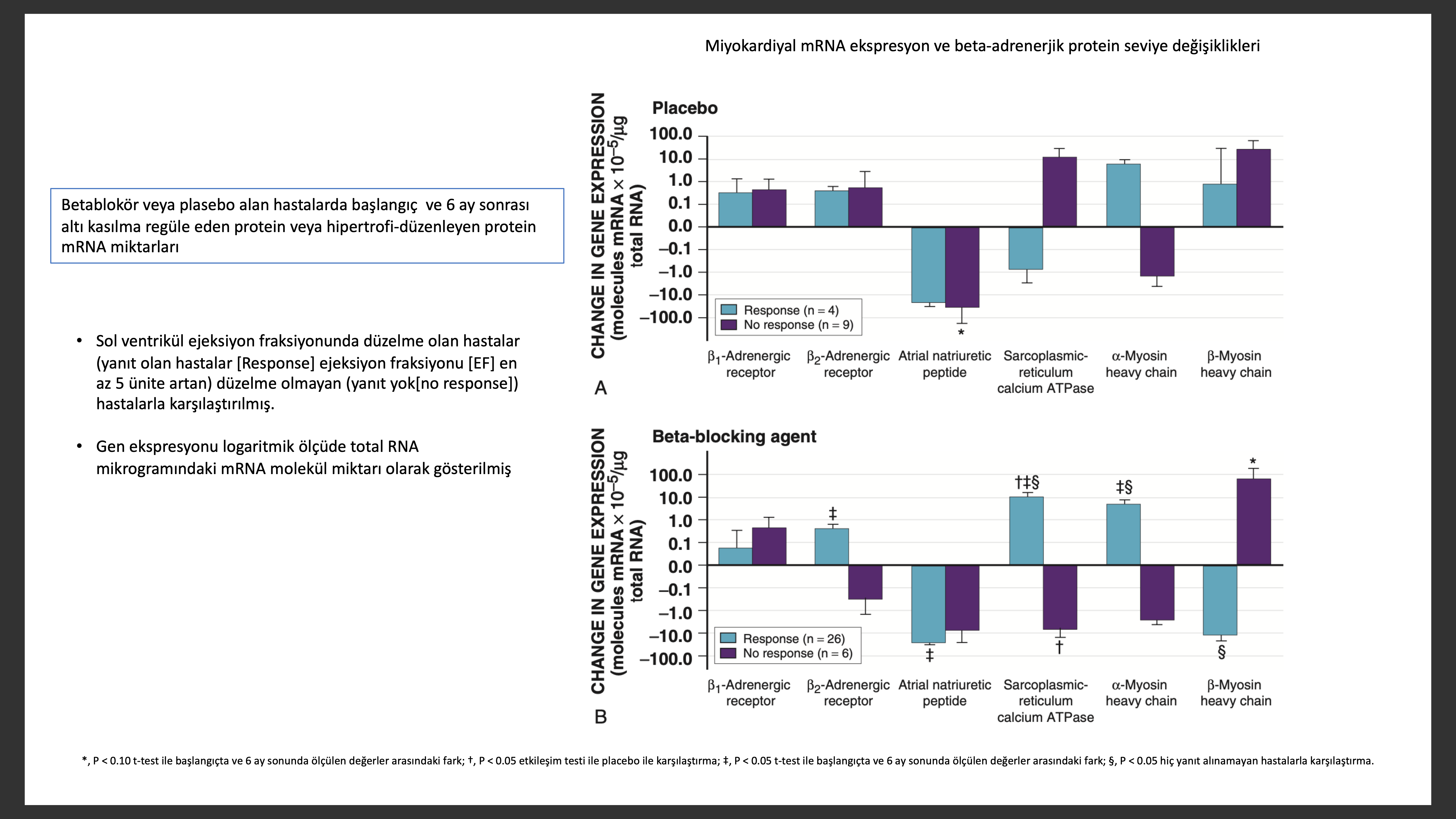
Kontraktil ve Düzenleyici Protein Anormalikleri
Eski çalışmalarda Kalp yetersizliğinden ölen hastaların kalplerinde miyofibriler ATP az aktivitesi azalmış olduğu gösterilmişti. Miyofibriler ATPaz, aktomiyozin ATPaz veya miyozin ATPaz aktivitelerinde azalma bazı hayvan kalp yetersizliği modelllerinde gösterildi. Kardiyak hipertrofi ve yetersizliğinde yapılan sonraki çalışmalarda ATP az aktivitesindeki anormallikler miyozin ağır zincir (MHC) in fetal izoformuna dönüşmesine bağlandı. Kobaylarda baskın MHC yüksek ATPaz aktivitesi olan “hızlı” V1 izoformudur (alfa-MHC [MYHC6]). Kobaylarda basınç-bağımlı hipertrofi veya MI sonrası düşük ATPaz aktivitesi olan “yavaş” V3 fetal MHC izoform (beta-MHC[MYHC7]) ekspresyonu artar ve V1 ekspresyonu azalır. Bu bulguları direkt olarak insan kalp yetersizliğine uyarlamak insanda predominant MHC izoformu yavaş V3 (MYHC7) olması sebebi ile zor olur. Normal insan miyokartında yapılan polimer zincir reaksiyon (PCR) çalışmalarında MYHC6, MHC RNA % 33 ünü oluşturmakta ancak yetersiz olan kalpte bu oran % 2 ye düşmektedir. Beta blokör alan sol ventrikül performansı düzelen hastalardan kalp biyopsisi alındığında mRNA MYHC6 seviye artışı, MYHC7 seviye azalması ve MYHC6/MYHC7 oran artışı görülür. Bu değişiklikler sol ventrikül performansı düzelmeyen hastalarda görülmez. MYHC6 ekspresyonu azalması dilate kardiyomiyopati (DCM) patofizyolojisinde önemli rol oynar.
Diğer bir kontraktil bozukluğa yol açan önemli değişiklik kontraktil protein olan miyoflamanların proteolizidir (miyositolizis). İleri kalp yetmezlik hastalarında görülen kardiak dekompansasyonda önemli bir katkı, alınan biyopsi örneklerinde hücre başına düşen miyofibril volümündeki belirgin azalmadır.
Miyoflaman düzenleyici miyozin hafif zincir, troponin-tropomiyozin kompleksi, ve titin gibi proteinlerin ekspresyonu ve/veya aktivitesi, kalp yetersizliğinde kontraktil fonksiyonu belirlemede önemli bir mekanizma olarak ileri sürülmüştür. Mekanik yüklenmeye maruz kalan hastaların atriyum ve ventriküllerinde miyozin hafif zincir izoform değişiklikleri görülür. Kalp yetersizliğinde troponin miktar ve/veya TnI ve TnC izoformlarında değişiklik olmamasına rağmen TnT izoformuna dönüşüm rapor edilmiştir. Normal yetişkin kalbinde TnT tek bir izoformda (cTnT3) bildirilmiştir. Son dönem kalp yetersizliği miyokart örneklerinde fetal cTnT1 ve cTnT4 ekspresyonu belirgin olarak artmıştır. Bu miyokart aktif geriliminde azalmaya sebep olabilir. Kalp yetersizliğinde titin izoformlarından postnatal sert olan N2B yerine fetal esneyebilen N2BA ya geçerek kalpte kompleans artışı bildirilmiştir.
Sitoskeletal Protein Anormallikleri
Kardiyak miyozitlerin sitoiskeleti aktin, orta filaman desmin, sarkomerik protein titin ve polimerizasyon ile mikrotübülleri yapan alfa ve beta tubulinden oluşur. Vinculin, talin, distrofin ve spektrin membran-bağlı proteinler olarak ayrı bir gurup oluşturur. Deneysel bazı çalışmalarda sitoiskelet ve membran-bağlı proteinlerin kalp yetersizliği patogenezinde rol aldıkları gösterilmiştir. DCM li hastalarda titin azalmıştır, sitoiskelet proteini desmin ve membran-bağlı proteinler (vinculin ve distrofin gibi) artmıştır. Distrofin proteininin proteolitik sindirimi kalp yetersizliğinde geri dönebilen sebeplerden biridir. Sitoiskelet ve onun sarkolema ile sarkomere ve ekstra sellüler matrikse bağlantısının kaybı miyosit ve miyokardiyal seviyesinde kasılma kusuruna sebep olur.
Beta-Adrenerjik Desensitization
Kalp yetersizliği olan hastalardan elde edilen ventrikül örneklerinde beta -adrenerjik reseptör yoğunluğunda, izoproterenole-bağlı adenyl siklaz uyarımında ve beta adrenerjik agonistler ile olan kontraktil yanıtta belirgin azalma olur. Beta adrenerjik reseptör azaltma düzenlemesi muhtemelen reseptör civarı NE seviye artışı ile ortaya çıkar. DCM hastalarında reseptör yoğunluk azalması öncelikle beta1-adrenerjik reseptör protein ve mRNA dır ve kalp yetersizliği ciddiyeti ile orantılı olarak azalır. Tersine beta2-adrenerjik reseptör protein ve mRNA sı değişmez veya artar. Ek olarak yetersiz kalpte GPCR ailesinden beta adrenerjik reseptör kinaz 1 (bARK1 , aynı zamanda G protein-eşleşen reseptör kinaz2 [GRK2] ismini de alır) ekspresyonu artar. Daha önce anlatıldığı üzere bARK1 beta1- ve beta2- adrenerjik reseptör sitoplazmik kıvrıntılarını fosforilize eder ve bu reseptörlerin kıvrıntı proteini beta-arrestine ilgisini artırır. Beta-arrestinin beta reseptör sitoplazmik kuyruğuna bağlanarak reseptörü heterotrimerik G proteinden ayırmakla kalmayıp aynı zamanda hedef reseptörleri klatrin-kaplı veziküllerin içerisine girmeye zorlar. Bu içeriye alış reseptör defosforilasyonunu teşvik eder ve beta adrenerjik reseptörün yüzeye çıkıp tekrar aktive olmasını sağlarken bir noktada reseptör endositoza uğrayıp geri dönüşüme girmez, lizozomlara iletilerek parçalanır. Bundan dolayı artmış bARK1 (GRK2) aktivitesi hem beta1 hem de beta2- adrenerjik reseptörlerini kalp yetersizliğinde duyarsızlaştıra bilir (desensitization). Beta reseptörlerin duyarsızlaşmasının kalp yetersizliğinde hem yararlı hem de zararlı etkileri olabilir. Yetersiz kalpte sol ventrikül kontraktilitesini azaltarak zararlı olurken enerji açlığı olan miyokart da enerji tüketimini azaltıp adrenerjik etkiden miyokardı koruyarak ,duyarsızlaşma (desensitizasyon) olumlu etki gösterebilir. Kalp yetersizliğinde lenfosit GRK2 protein seviyesinin kardiyovasküler mortalitenin bağımsız göstergesi olduğu ve demografik ve klinik değişkenlere ek olarak prognostik ve klinik değeri olduğu gösterilmiştir.
Miyokardiyumdaki Değişiklikler
Yetersiz kalpteki değişiklikler kardiyak miyozit volümünde olan ve ekstrasellüler matriksin volüm ve içeriğinde olanlar şeklinde kategorize edile bilir. Gün geçtikçe artan deliller göstermektedir ki, miyokartdaki miyozit içeriği ilerleyici kaybı nekrotik, apoptotik veya otofajik hücre ölüm yolakları yolu ile olmakta ve ilerleyici kalp fonksiyon bozukluğu ve sol ventrikül yeniden yapılanmasına yol açmaktadır.
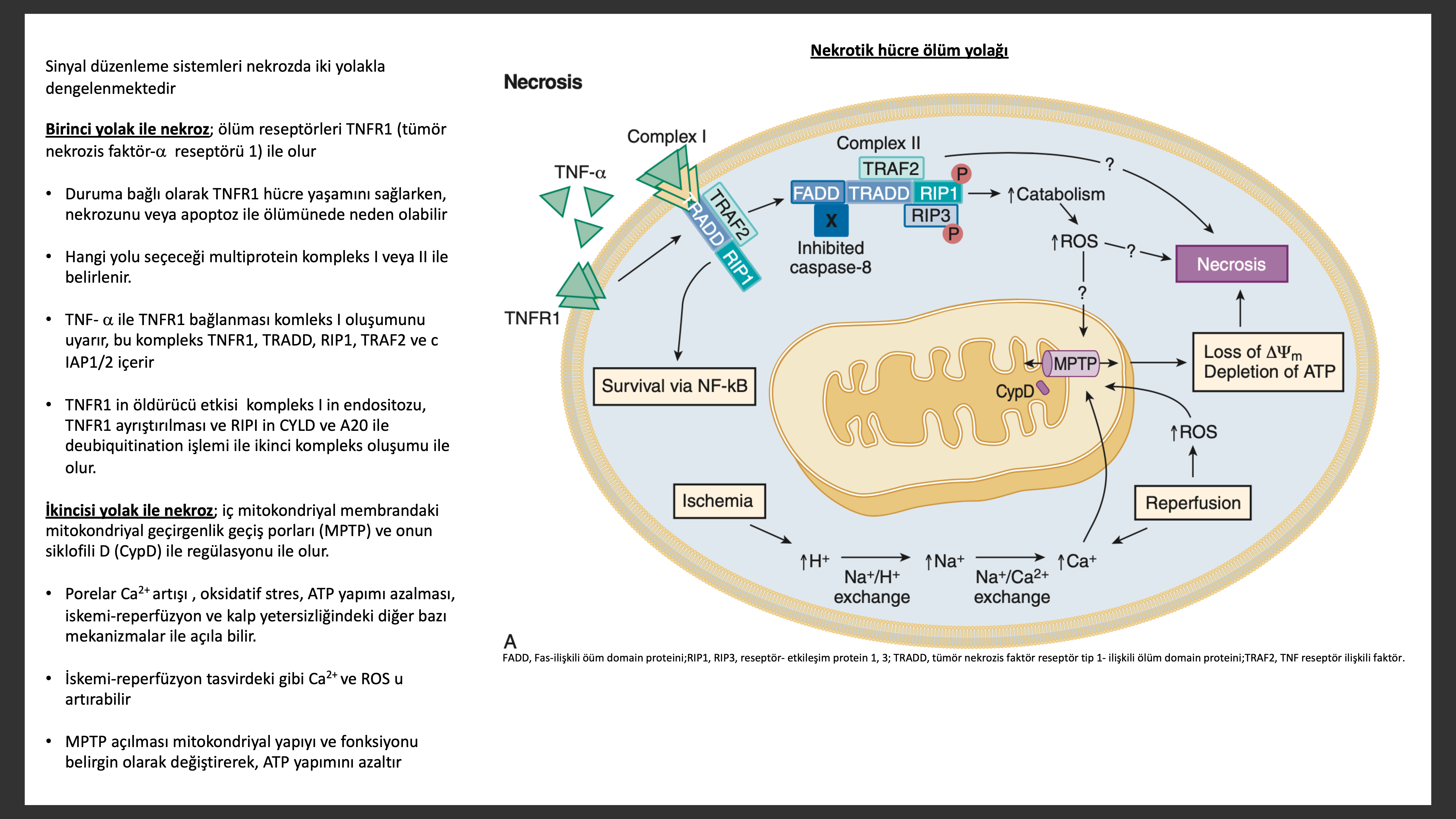
Nekroz
Önceleri nekrozun hücre ölümü “pasif” formu olduğu düşünülürken gün geçtikçe artan bulgular nekrotik hücre ölümünün de “kontrollü” olduğunu bize göstermektedir. Kalpte düzenlenmeyen ve düzenlenen nekrotik ölümün ne oranda olduğu halen bilinmiyor; ancak MI, kalp yetersizliği ve serebrovasküler olay sonrası nekrozun önemli bir bölümünün düzenlenen nekroz olduğu bilinmektedir. Plazma bütünlüğünün kaybolması, hücresel adenozin trifosfat (ATP) kaybı nekrozun karakteristikleridir. Plazma membran fonksiyon bozukluğu hücrelerin şişme ve patlamasına sebebp olur. Ayrıca mitokondri gibi organellerde şişerler. Kalpte artan plazma membran geçirgenliği Ca2+ un hücre içine sızmasına, kontraktil proteinlerin bu aktivatöre yoğun maruz kalmasına, sonuçta miyoflamanlar arası aşırı etkileşim (kontraksiyon bantları arası)ve hücresel membranın daha fazla bozulması şeklindedir. Nekrotik hücre ölümü iskemik kalp hastalığında, miyokart zedelenmesinde, toksine (ör daunorubisin) maruz kalmada, enfeksiyon ve inflamasyonda olabilir. Nörohumoral aktivasyonda nekrotik hücre ölümüne sebep olabilir. Örneğin miyokart dokusundaki NE konsantrasyonları, aynı zamanda kalp yetmezliğinde dolaşımdaki NE, deneysel modellerde miyokart nekrozu gelişmesi için yeterlidir. Anjiyotensin II, ET veya TNF aşırı miktarları yine deneysel modellerde miyokart nekrozuna sebep olmuştur.
Apoptozdan farklı olarak nekrozda hücre membranlarının yarılması , tehlike içeren moleküler yollar (danger-associated mollecular patterns [DAMPS] denilen hücre içeriğinin dışarı salınarak granülosit, makrofaj ve kollajen salan fibroblast gibi yoğun inflamatuar yanıt oluşturacak hücrelerin zedelenme alanına gitmesine sebep olur. Son ürün miyokartda yapısal ve fonksiyonel içerikleri bozan fibrotik skar dokusu oluşmasıdır. Bugüne kadar çalışılmış düzenlenen hücre ölüm yolakları TNF sinyalizasyonu ile tip 1 TNF reseptörü (TNFR1) üzerinden ve iç mitokondial membrandaki mitokondrial geçirgenlik geçiş porları (MPTP) açılması ile iç mitokondrial membran elektriksel potansiyel farkı kaybı (DYm) ile ATP tükenmesidir.
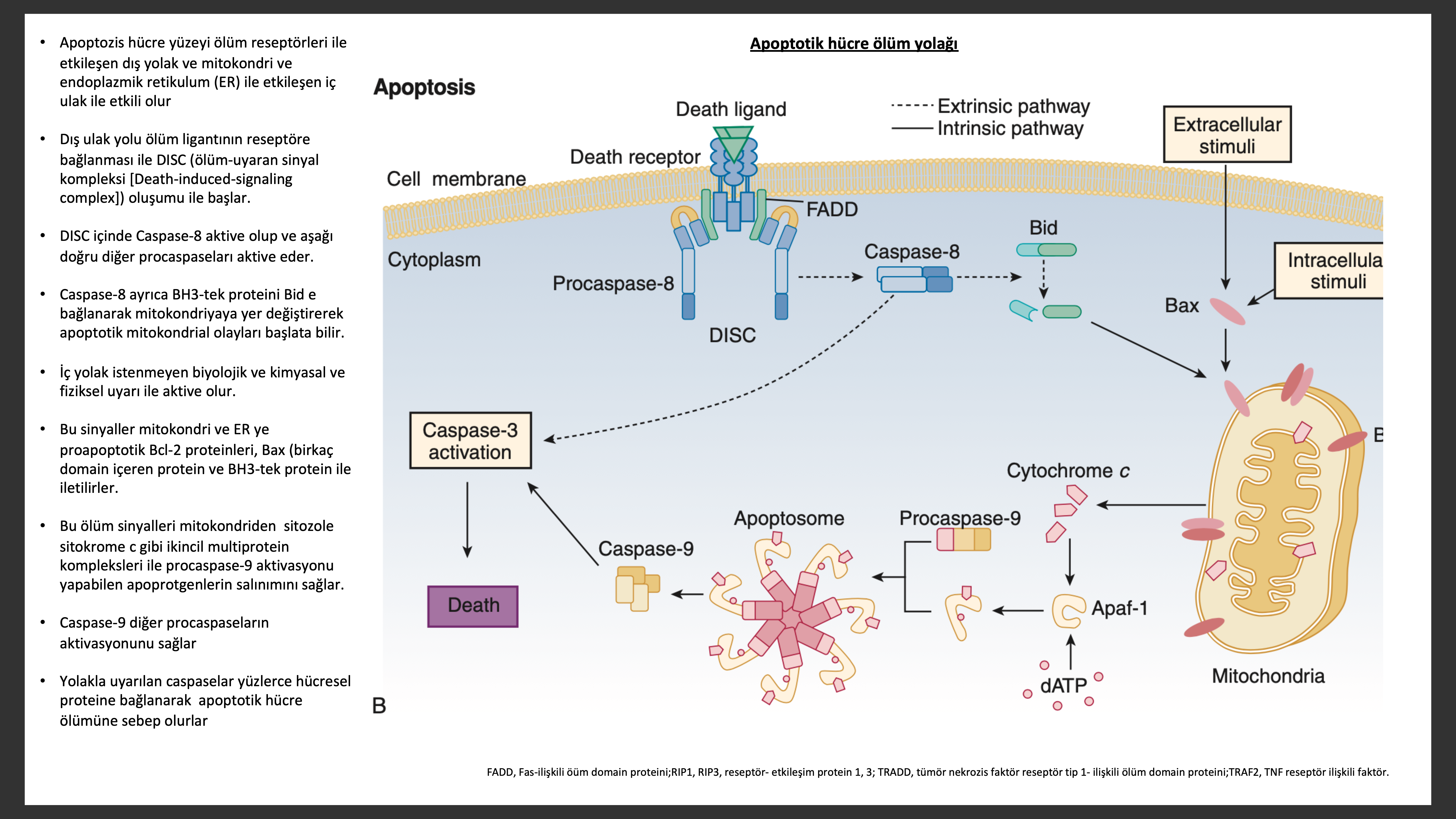
Apoptozis
Apoptozis veya diğer adı ile programlı hücre ölümü, ileri derecede organize bir intihar programı ile çok hücreli organizmalarda hücrelerin ortadan kaldırıldığı, koruyucu evrimsel mekanizmadır. Apoptozis iki aracı mekanizma ile gerçekleşir. Dış yolak hücre reseptörlerini, iç yolak ise mitokondri ve endoplazmik retikulumu (ER) kullanarak etki gösterir, her iki yolakta caspaseı aktive eder. Her iki yolak arasındaki bağlantı sinyalleri artırarak öldürme etkinliğini artırır. İç yolak uygunsuz besinler, veya hayatta kalma faktörleri, hipoksi, oksidatif stres, besin stresi, proteotoksik stres, DNA zedelenmesi ve kimyasal ile fiziksel toksinler gibi apoptoz uyarılarının iletisinden sorumludur. Bu uyarı mitokondriden sitokrom c gibi apoptojenik proteinlerin ve ER den lümene Ca2+ salınmasına neden olur. Gelişme sürecinde ve postnatal hayatta zarar görmüş veya transforme olmuş hücrelerin kontrolü ile hemostazın ve dengenin sağlanmasında apoptozis çok önemlidir. Ancak akut iskemi ve DCM gibi patolojik durumlarda apopotik program uygunsuz şekilde tetiklenerek, hücre ölümü ile organ yetersizliğine sebep olabilir. Nekrozdan farklı olarak hücreler apoptozisde şişmez tersine çeker ve kısalır ve membran ile çevrelenmiş fragmanlara parçalanırlar. İçerde apoptotik cisim (bodies) denilen yoğunlaşmış kromatin materyal parçaları içerir. Apoptotik süreç geç evrelerine kadar membran bütünlüğünün korunması ölen hücrelerin makrofajlar tarafından yutularak reaktif hücre içeriklerin salınmasını dolayısı ile de inflamatuar reaksiyonu engeller.
Yetmezlik olan kalpte miyosit apoptozu gösterilmiştir. İn vitro olarak beta1-adrenerjik reseptör üzerinden etkili katekolaminlerin, anjiyotensin II, ROS, inflamatuar sitokinlerin (ör TNF) ve mekanik gerginleşme gibi kalp yetmezlik patogenizinde suçlanan faktörlerin apoptozu tetiklediği gösterilmiştir. Transgenetik farede iç veya dış yolaktan herhangi biri aktive edildiğinde sol ventrikül dilatasyonu ve dekompansasyon oluşmaktadır. Ancak yetmezlik olan kalpteki miyozitde apoptozun esas miktarını tespit etmedeki belirsizlik kalp yetersizliğinde apoptozun tam olarak fizyolojik önemini ve sonuçlarını anlamamızı zorlaştırmaktadır. Klinik ve deneysel verilerin tümüne bakıldığında apoptozun kalp yetersizliğinde önemli rol oynadığını düşünebiliriz.
Otofaji
Otofaji, organalleri, protein ve lipitleri hücre içi çift membranlı vezikülde (otofagozom) tecrit ederek içerikleri parçalanmak için lizozoma iletilen homeostatik hücresel işlemdir. Nekroz ve apoptozisden farklı olarak otofaji hücre içi protein ve organel kalitesini ve miktarını ayarlayan önemli bir hayatda kalma mekanizmasıdır. Makrootofaji, mikrootofaji ve chaperon -bağımlı otofaji olmak üzere üç tür otofaji vardır. Genellikle aksi söylenmez ise otofaji terimi makrootofaji için kullanılır. Otofaji ile tüm hücre parçalanırsa otofajik hücre ölümü denir. Son çalışmalar hipertrofik, yetersiz ve hiberne miyokartda otofajik hücre ölümünü göstermişlerdir. Kalp yetmezliği kalp örneklerinde otofajik hücre ölümü % 0.3 civarındadır, ancak basınç yükü olan kalplerde baskın olan hücre ölümü otofaji ve onkozisdir. Son çalışmalar otofajinin kalpte fizyolojik rolü olduğunu ve istenmeyen etkilerin otofajiden çok otofagozomların (azalmış otofajik akım) temizlenmesinin azalmasından kaynaklandığı düşünülmektedir.
Nekroz ve apoptoz arasındaki ayrım bazı koşullarda belirgin olsa bile kalp yetersizliğinde ayrım çok net değildir. Benzer mekanizmalar her iki tür hücre ölümünde etkili olabilir. Farklı hücre ölümleri yerine devam eden hüvre ölümleri senaryosu ilerleyici miyozit kaybı ve hastalık ilerlemesinin temelini oluşturur.
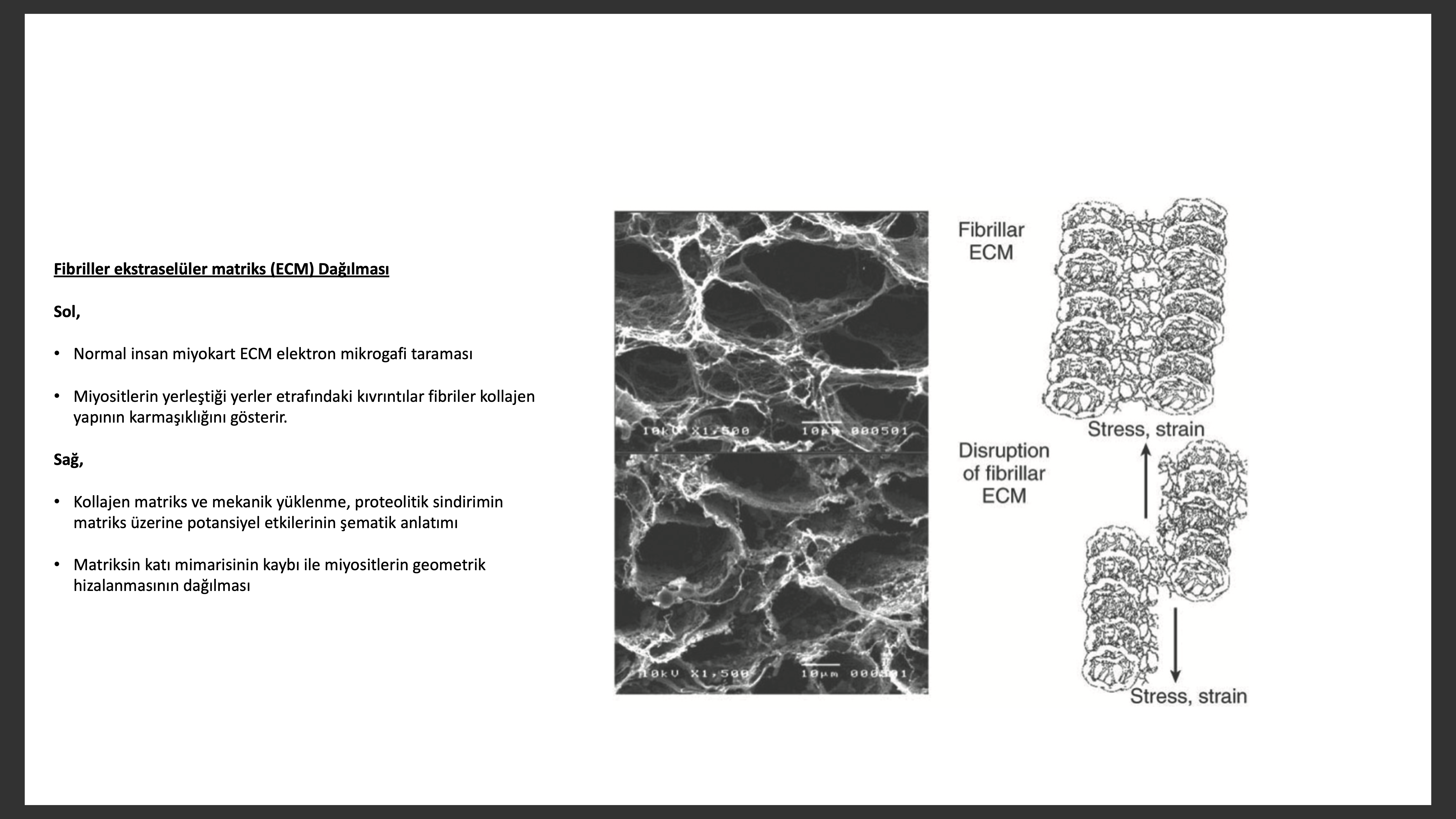
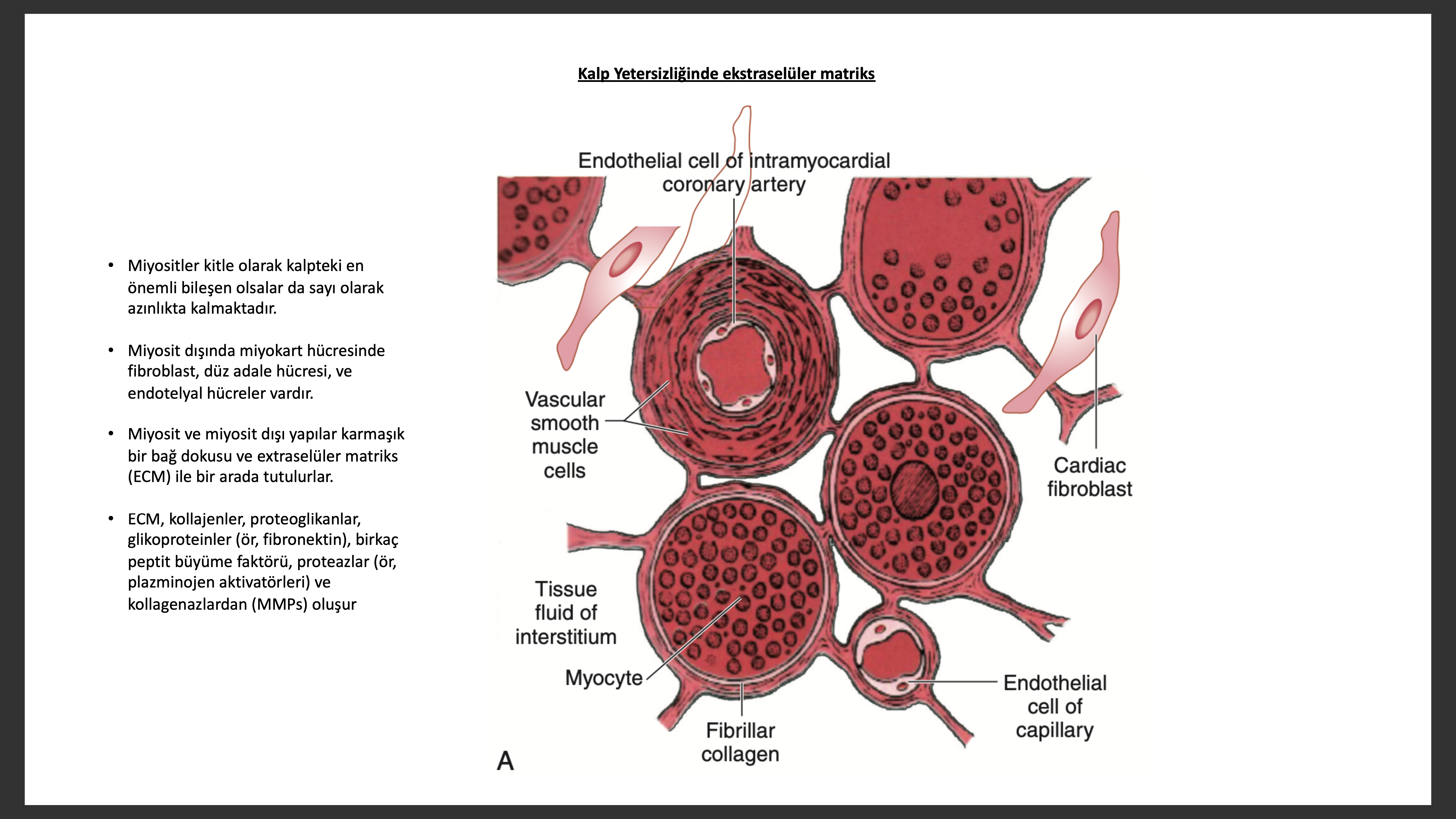 Yeniden yapılanma sırasında sırasın da ikinci önemli miyokardiyal adaptasyon ekstra cellüler matriksde (ECM) olur. ECM temel membran, miyositleri saran fibriler kollajen ağı, proteoglikanlar, glikozaminoglikanlar ve matrisellüler proteinler gibi özel proteinlerden oluşur. Kalpteki majör fibriler kollejenler Tip I ve III dür, Tip I/III oranı 1.3/1 ile 1.9/1 arası bir orandadır. Miyokardiyal fibriler tip I ve III kollajen, bitişik miyositlerin bütünlüğü ve miyofibrillerin hizalanması için integrin ve sitoskeletal proteinler ile etkileşime girerler.
Yeniden yapılanma sırasında sırasın da ikinci önemli miyokardiyal adaptasyon ekstra cellüler matriksde (ECM) olur. ECM temel membran, miyositleri saran fibriler kollajen ağı, proteoglikanlar, glikozaminoglikanlar ve matrisellüler proteinler gibi özel proteinlerden oluşur. Kalpteki majör fibriler kollejenler Tip I ve III dür, Tip I/III oranı 1.3/1 ile 1.9/1 arası bir orandadır. Miyokardiyal fibriler tip I ve III kollajen, bitişik miyositlerin bütünlüğü ve miyofibrillerin hizalanması için integrin ve sitoskeletal proteinler ile etkileşime girerler.
 Matriselüler proteinler yapısal olmayan ECM proteinleridir, hücre yüzey reseptörleri, yapısal proteinler, büyüme faktörü ve sitokinler gibi çözünebilir ekstraselüler faktörler ile etkileşime girerek düzenleyici rol oynarlar. Osteopontin (OPN[Eta-1]) kardiyak miyosit, fibroblast ve miyofibroblast gibi değişik hücre türlerinde bulunan matriselüler proteindir. Bulunduğu yer ve moleküler yapısı sebebi ile , OPN muhtemelen hemodinamik yüklenme sonrası kardiyak yeniden yapılanmada, ECM ve kardiyak miyozitler arasındaki iletişimi sağlar. Hayvan modellerinde kardiyak hipertrofi ve yetmezlikte, miyokardiyal iskemi ve DCM olan kalp yetmezlik hastalarında OPN miktarı artmıştır. Kalp yetersizliği ciddiyeti ile direkt ilişkili olarakperiferik dolaşımda OPN artmıştır.
Matriselüler proteinler yapısal olmayan ECM proteinleridir, hücre yüzey reseptörleri, yapısal proteinler, büyüme faktörü ve sitokinler gibi çözünebilir ekstraselüler faktörler ile etkileşime girerek düzenleyici rol oynarlar. Osteopontin (OPN[Eta-1]) kardiyak miyosit, fibroblast ve miyofibroblast gibi değişik hücre türlerinde bulunan matriselüler proteindir. Bulunduğu yer ve moleküler yapısı sebebi ile , OPN muhtemelen hemodinamik yüklenme sonrası kardiyak yeniden yapılanmada, ECM ve kardiyak miyozitler arasındaki iletişimi sağlar. Hayvan modellerinde kardiyak hipertrofi ve yetmezlikte, miyokardiyal iskemi ve DCM olan kalp yetmezlik hastalarında OPN miktarı artmıştır. Kalp yetersizliği ciddiyeti ile direkt ilişkili olarakperiferik dolaşımda OPN artmıştır.
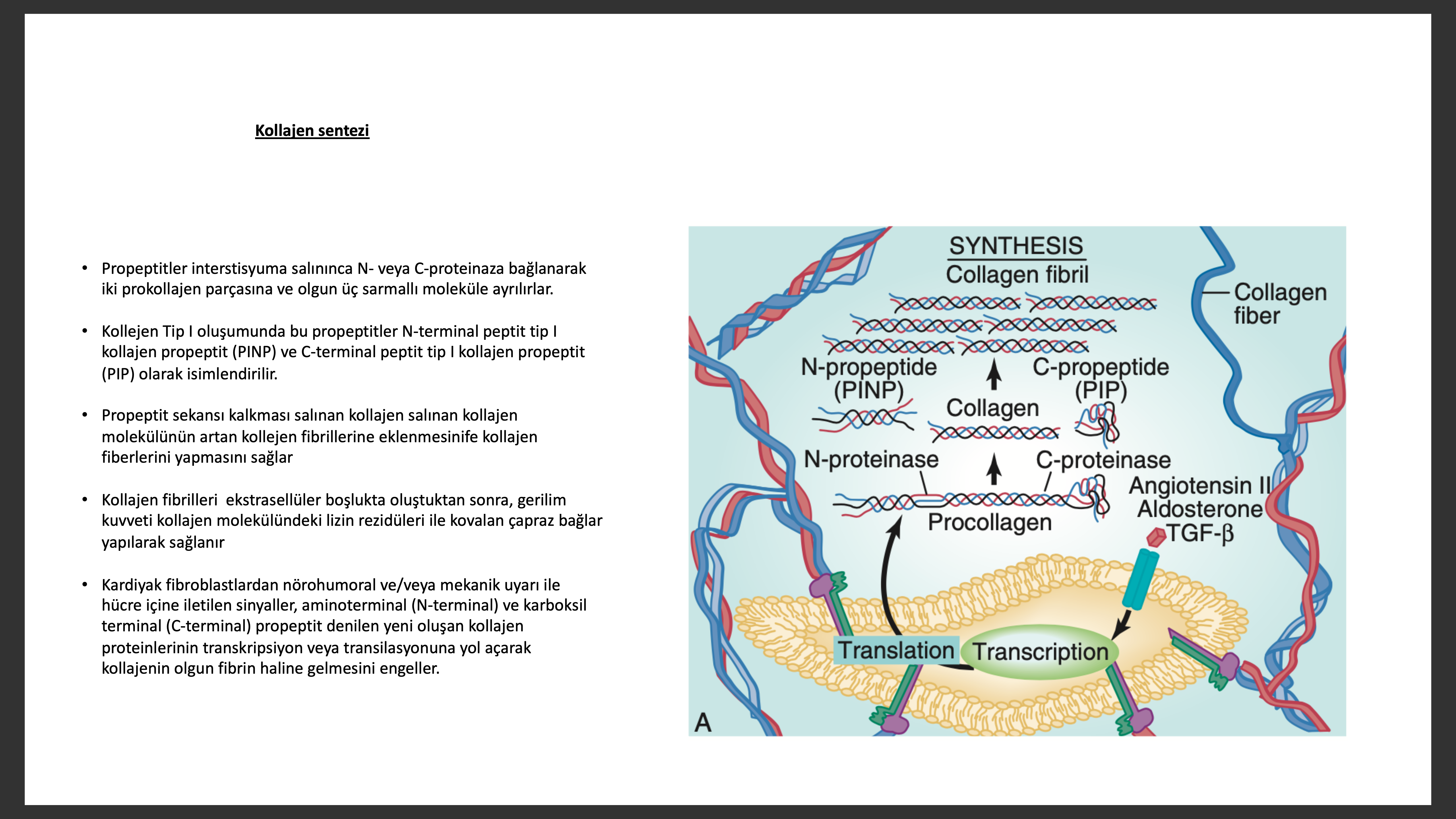
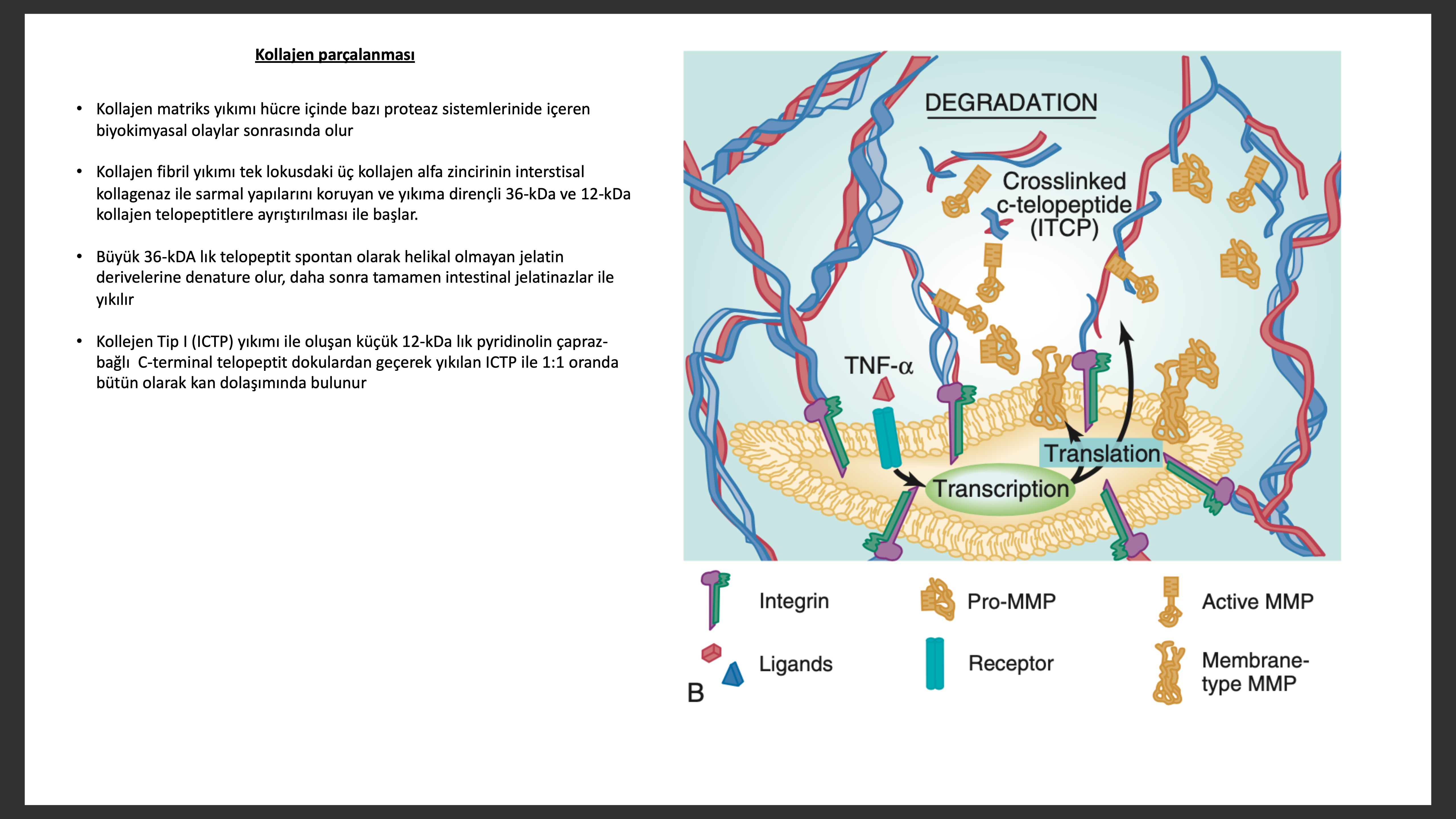
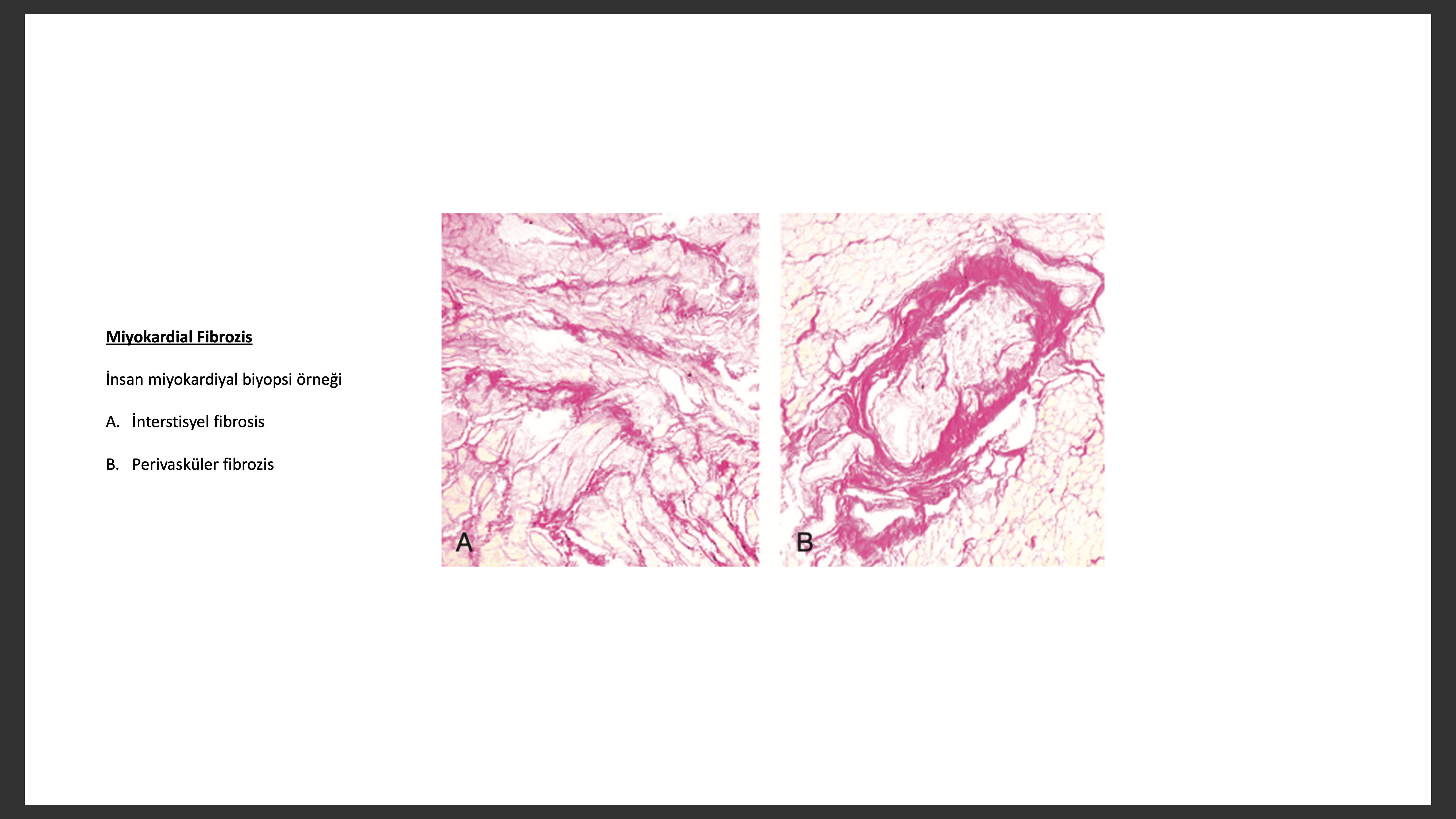
Kardiyak yeniden yapılanma sırasında fibriller kollajen sentez değişiklik ve bozuklukları, kollejen çapraz bağlama derecesi ve teker teker kardiyak miyositleri bağlayan kollejen destek yapı kaybı gibi ECM de önemli değişiklikler olur. Kollajen geri dönüş belirteçlerinin yaş karşılaştırmalı guruplara göre DCM de arttığı gözlenmiştir. İdiyopatik veya iskemik DCM hastalarında serum N-terminal peptit tip III kollajen propeptit (PIIINP) seviyelerinin mortalitenin bağımsız belirteci olduğu gösterilmiştir. Rales çalışmasında spirinolakton ile tedavi edilen hastalarda C-terminal peptit tip I kollajen propeptit (PIP) ve PIIINP serum seviyeleri düşüp placebo gurubunda düşmemiş olması aldesteronun ECM sentezinde önemli rol oynuyor olabileceğini düşündürmüştür. Yine ECM nin üç boyutlu yapısının kalp yapısını düzenlemede ve fonksiyonda önemli rol oynadığı düşünülmektedir.
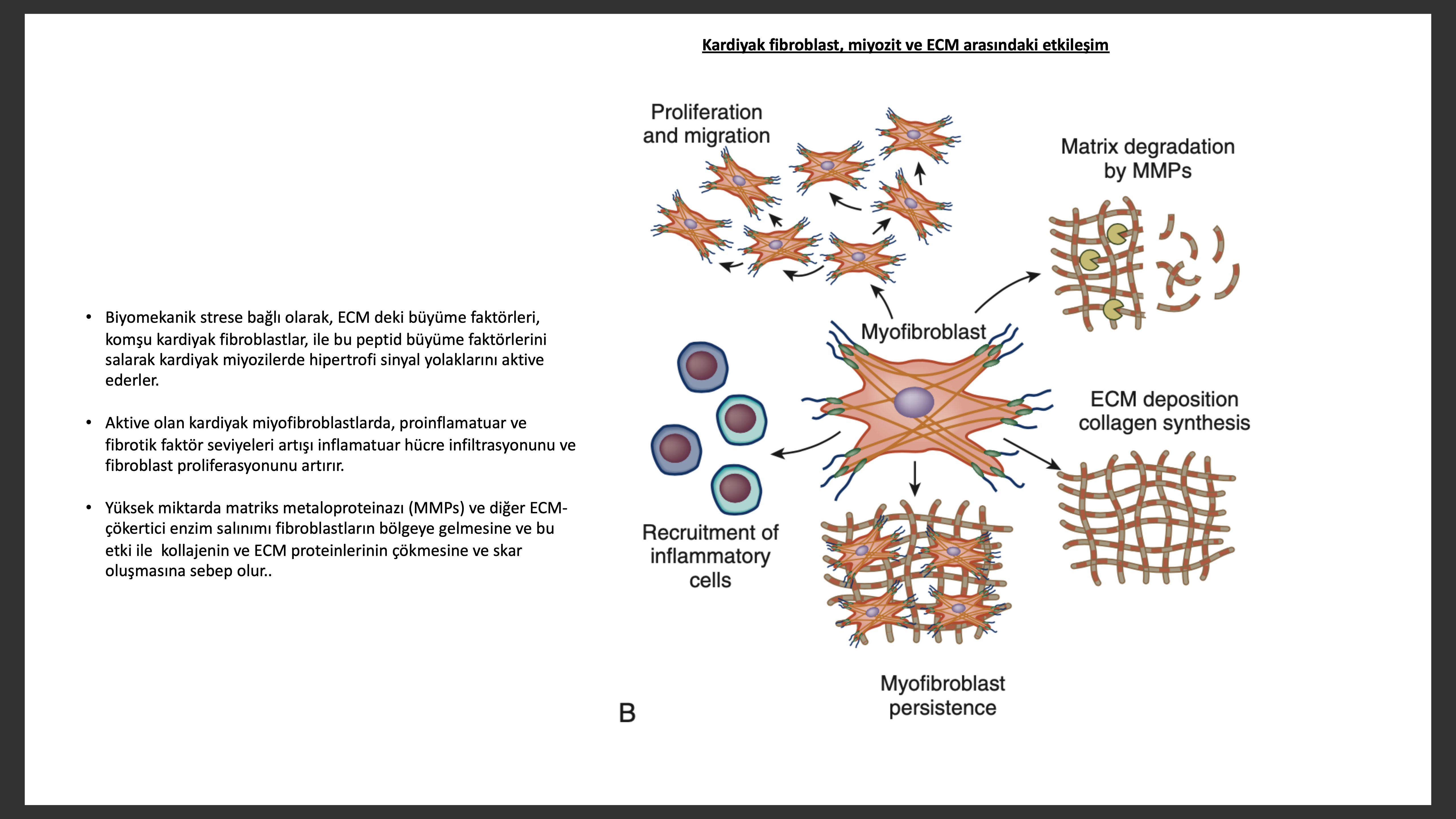
Kardiyak Fibroblast ve Mast Hücreleri
Kalpteki miyosit dışı hücrelerin % 90 nı oluşturan fibroblastların birincil görevi, kollajen I,III,IV, laminin ve fibronektin gibi ECM içeriğinin salınımıdır. Mekanik stress ve nörohumoral uyarana yanıt olarak bir gurup fibroblast fenotipik olarak miyofibroblasta değişerek karakteristik olarak a-düz adele aktin ekspreyonunu ve salınım aktivitesini artırır. Son çalışmalar kollajen salınması ve sonrası oluşan kollajen fiber kontraksiyon/hizalama sorumlusu miyofibroblastların, doku zedelenmesi sonrası aktive olan lokal doku fibroblastlarından köken aldığını göstermektedir. Miyofibroblastlar doku etrafına göç ederek skar oluşmasında önemli rol oynarlar. Kardiyak miyofibriblastlar birçok parakrin sinyal yolu ile kardiyak miyozit fenotipinde önemli rol oynarlar. Bazı kanıtlar kardiyak fibroblast ve miyozitlerin etraftaki hücreleri düzenleyen proteinler salgıladığını göstermektedir. Şimdiye kadar transforming growth factor-b1(TGF-b1), fibroblast growth factor-2 (FGF2), IL-6 aile üyeleri ve yeni keşfedilen sitokin IL-33 ilgili olduğu bulunana proteinlerdir. Kanıtlara göre miyokartı yuva edinip yerleşen kemik iliği menşeyli mast hücreleri ECM yeniden yapılanmasında önemli rol oynarlar. Esasen kan damarları ve miyositler arasında lokalize olan miyokardiyal mast hücreleri, profibrotik sitokin ve büyüme faktörü salma kabiliyetleri sayesinde ECM yeniden yapılanmasını etkilerler. Deneysel çalışmalarda inflamasyon sırasında eklenen mast hücreleri, TGF-b1 ile fibroblast aktivasyonundan, miyokardiyal fibroz ve diastolik fonksiyon bozukluğunun sorumlusudur.
Daha öncede söylendiği gibi ilerleyen kalp yetersizliğinin histolojik imzalarından bir tanesi kalbin giderek artan kollejen içeriğidir (miyokardiyal fibrozis). Yetersiz insan miyokartındaki çalışmalar kollajen tipI, III, VI, IV, fibronektin, laminin ve vimentim miktarlarında artış ve iskemik kardiyomiyopatili hastalarında tip I/III kollajen oranı azalır. Klinik çalışmalar yetersiz kalpte ilerleyici kollajen çapraz bağ kaybı ile birlikte her bir miyosit kollajen ağ iletişim kaybının sol ventrikül yapı ve fonksiyonunu etkilediği düşünülmektedir. Dahası fibriler kollajenin çapraz bağ kaybının miyokart zedelenmesi sonrası ilerleyici sol ventrikül dilatasyonuna sebep olduğu düşünülür. Kollajen birikimi reaktif temelde, miyokart ölümü şart olmadan intramural koroner arterler, arterioller (perivasküler fibrozis) , ve intersisyel alanda (interstisyal fibrozis) olabilir. Alternatif olarak kollajen kardiyak miyosit hücre nekrozuna yanıt olarak oluşan mikroskobik skar dokusu sonucunda oluşa bilir. Bu skarlaşma veya “ fibroza homojen olmayan aktivasyon, dal bloğu ve disenkroni ile atrial ve ventriküler aritmiler ve ani kardiyak ölüm için yapısal zemin hazırlar. Fibroblastı aktive eden molleküllerin hepsini bilmemekle birlikte kalp yetersizliğinde salınan çoğu klasik nörohormon (ör, anjiyotensin II, aldesteron) ve sitokinler (ET, TGF-b, cardiotrophin-1) fibroblastı aktive eder.
Fibriler kollajen matriksin önceleri göreceli olarak statik bir kompleks oluşturduğu düşünülsede, artık bu yapısal proteinlerin hızlı dönüşüme girdiği kabul edilmektedir. Kardiyak yeniden yapılanmanın patogenezini anlamamızdaki önemli bir gelişme kollagenolitik bir enzim ailesi olan ve yetmezlik olan kalpte bulunan matriks metalloproteinazlarının (MMPs) keşfidir. Konsept olarak ECM bozulmasının miyosit kümelerini yeniden hizalayarak sol ventrikül dilatasyonu ve duvar incelmesi ve senkronize olmayan sol ventrikül kontraksiyonu ile sol ventrikül dilatasyonuna yol açması beklenir. MMPs nin aktivasyonunu tetikleyen mekanizmalar tam olarak bilinmemekle birlikte, TNF ve diğer sitokinlerin ve yetersiz kalpte salınan peptit büyüme faktörlerinin MMPs yi aktive etmede yeterli olacağı düşünülür. Kalp yetersizliğinde matriks yeniden yapılanmasının biyolojisi sadece MMP aktivasyonu olup, olmamasından daha komplekstir, çünkü matriks bozulması, doku inhibitör matriks metaloproteaz (TIMP) glikoproteinler tarafından da kontrol edilir. TIMP ler kalbin kollajen matriksinin parçalanmasına neden olan enzimlere bağlanıp parçalanmadan koruyarak MMP aktivasyonunu kontrol eder. TIMP ailesinin halen TIP-1, -2, -3 ve -4 üyeleri olup kalpte fibroblast ve miyositlerde bulunur. TIMP ler her türlü aktif MMP doğal inhibitörü olarak salınan proteinlerdir, ancak farklı üyeler farklı derecede MMP inhibisyonu yapar. Literatür MMP aktivasyonunun ilerleyici sol ventrikül dilatasyonu, TIMP ekspresyonun ise ilerleyici miyokart fibrozisi yaptığını ileri sürer.
Kodlama Yapmayan RNA
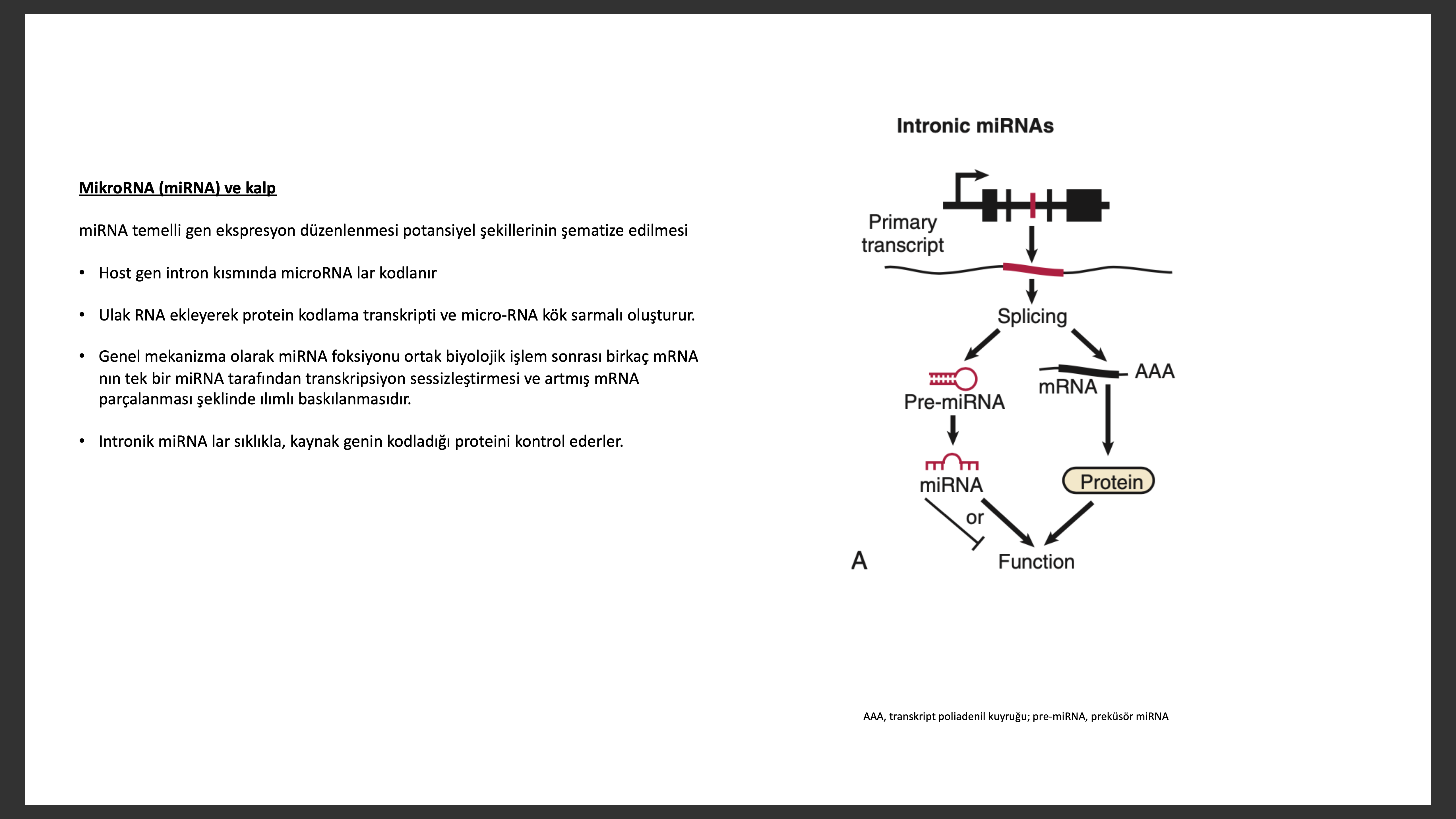
Bir zamanlar “transkripsiyon gürültüsü” ismi verilen kodlama yapmayan RNA lar, kalp yetersizliğinde potansiyel bir belirteç ve tedavi hedefi olarak gündeme gelmektedir. Genomun kodlama yapmayan kısmı aktif olarak kopyalanarak binlerce kısa düzenleyici ve uzun gen ağlarını düzenleye bilen kodlama yapmayan RNA lar oluştururlar. Kodlama yapmayan RNA lar uzunluklarına göre sınıflandırılırlar. Ufak kodlama yapmayan RNA kopyaları büyüklükte 200 nükleotitden daha ufak olan küçük etkileşen RNA (SiRNA) lar ve mikro RNA (miRNA) lardır. Büyüklüğü 200 nükleotitden fazla olan kopyalar ise uzun kodlama yapmayan RNA (lncRNA) lar ismini alır. MikroRNA lar hemen hemen bütün hücresel işlemlerde görev alırlar. Uzun lncRNA ise yine gen ve protein seviyelerini düzenler ancak bunu daha komplike ve farklı mekanizmalar ile yapar.
Deneysel çalışmalar kalpteki yeniden yapılanmada mikroRNA ların daha önemli etkisi olduğunu gösterilmiştir. Micro RNA lar özgül hedef RNA larla eşleşip çevrimi baskılama veya mRNA parçalama (gen susuturma) yoluyla etkiyi baskılarlar. MikroRNA bağlanma özgüllüğü birbirini tamamlama temelinde yaklaşık mikroRNA 5’ son 6 nükleotit (nt) bölgesi ile 3’ çevrilmemiş bölgesi (UTR) eşlenmesi ile olur. Mikro RNA ların benzer hedef mRNA ya bağlanması hedef genlerin baskınlığını azaltır. Her bir microRNA benzer fonksiyonları olan mRNA guruplarını uyararak karmaşık biyolojik işlemleri yönetir. Son deneysel kalp yetersizliği modellerinde microRNA ların istenmeyen veya patolojik yeniden yapılanmada görev alabileceği gösterilmiştir.
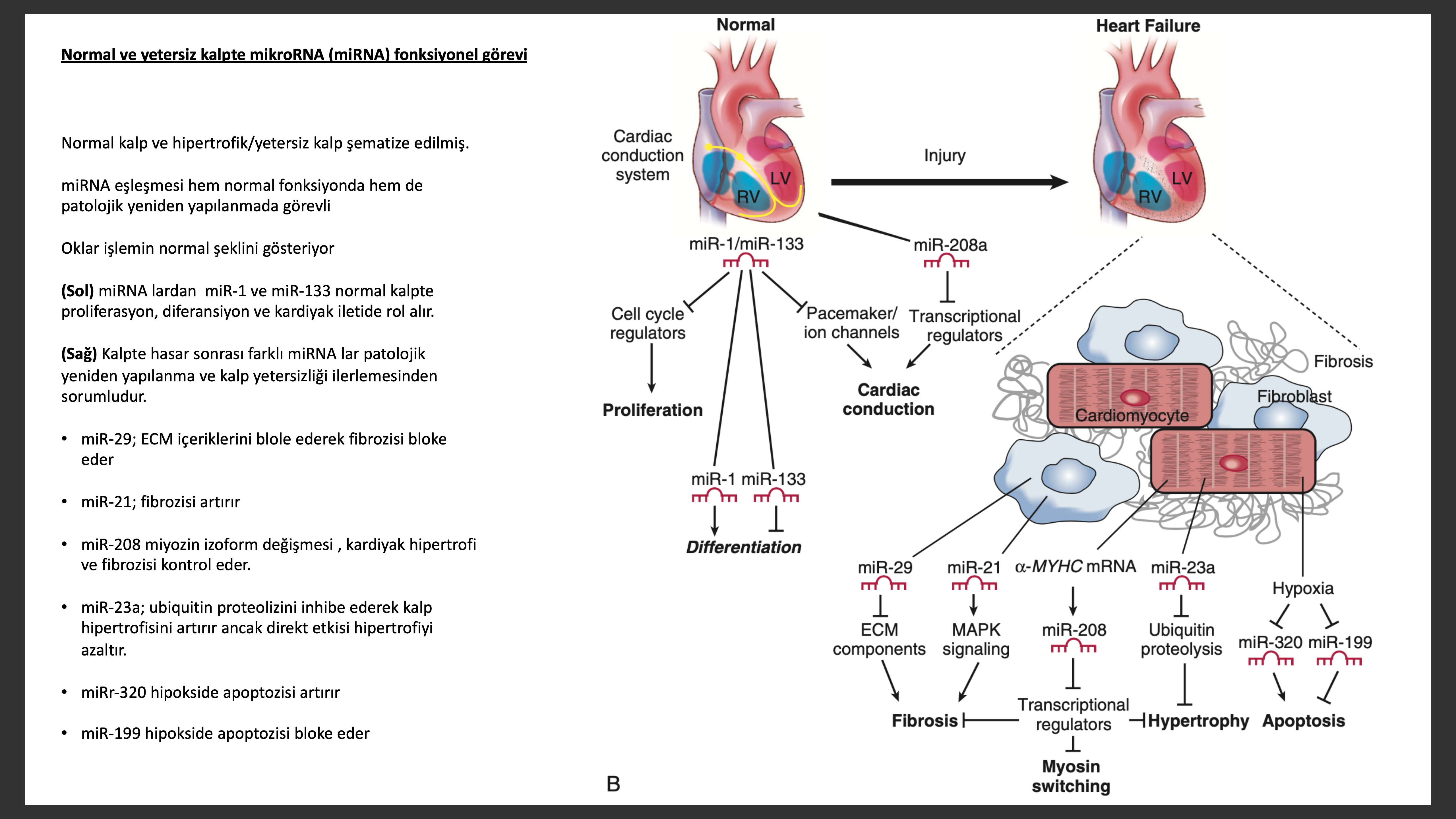
Buna göre microRNA kardiyak miyosit biyolojisi, hücre yaşamı, ECM yeniden yapılanması ve nörohumoral aktivasyonda önemli rol alan yapıları kontrol eder. Stres uyarıları ile koordine olarak artan microRNA ve bununla ayarlanan gen iletişimi “kalp yetersizliği fenotipini” belirleyerek , microRNA ların tek başına veya kombine olarak adapte ediciden patolojik kardiyak yeniden yapılanmaya geçişini kontrol ediyor olabilir. Dahası bazı microRNA lar tedavi hedefi haline gelebilir, kimyasal olarak modifiye edilen bazı özgül micro RNA ları hedef alarak özgül microRNA nın yine özgül mRNA ya bağlamasını engelleyebilir. Uzun, kodlama yapmayan RNA lar mekanik olarak micro RNA dan daha karmaşıktır ve genoma birkaç değişik seviyeden etki eder. Örneğin, lncRNA lar RNA lar, proteinler ve DNA ile etkileşerek molekülleri aktive edip veya sessizleştirerek yapısal değişiklik yapabilir. Son çalışmalar insanda kalp yetersizliğinde miyokardiyal lncRNA yapısının değiştiğini ve hemodinamik yüklenmede bazı lncRNA ların kardiyak yapı düzenlenmesinden ve fonksiyondan sorumlu olabileceğini göstermektedir.

Sol Ventrikül Yapısındaki Değişiklikler
Yetersiz miyosit ve miyokartdaki biyolojik değişiklikler geniş ölçüde kardiyak yeniden yapılanma sırasında olan ilerleyici sol ventrikül dilatasyonu ve disfonksiyonundan sorumludur. Sol ventrikül yeniden yapılanması sırasında olan yapısal değişikliklerin çoğu kalp yetersizliğinin ilerde kötüleşmesine sebep olacaktır
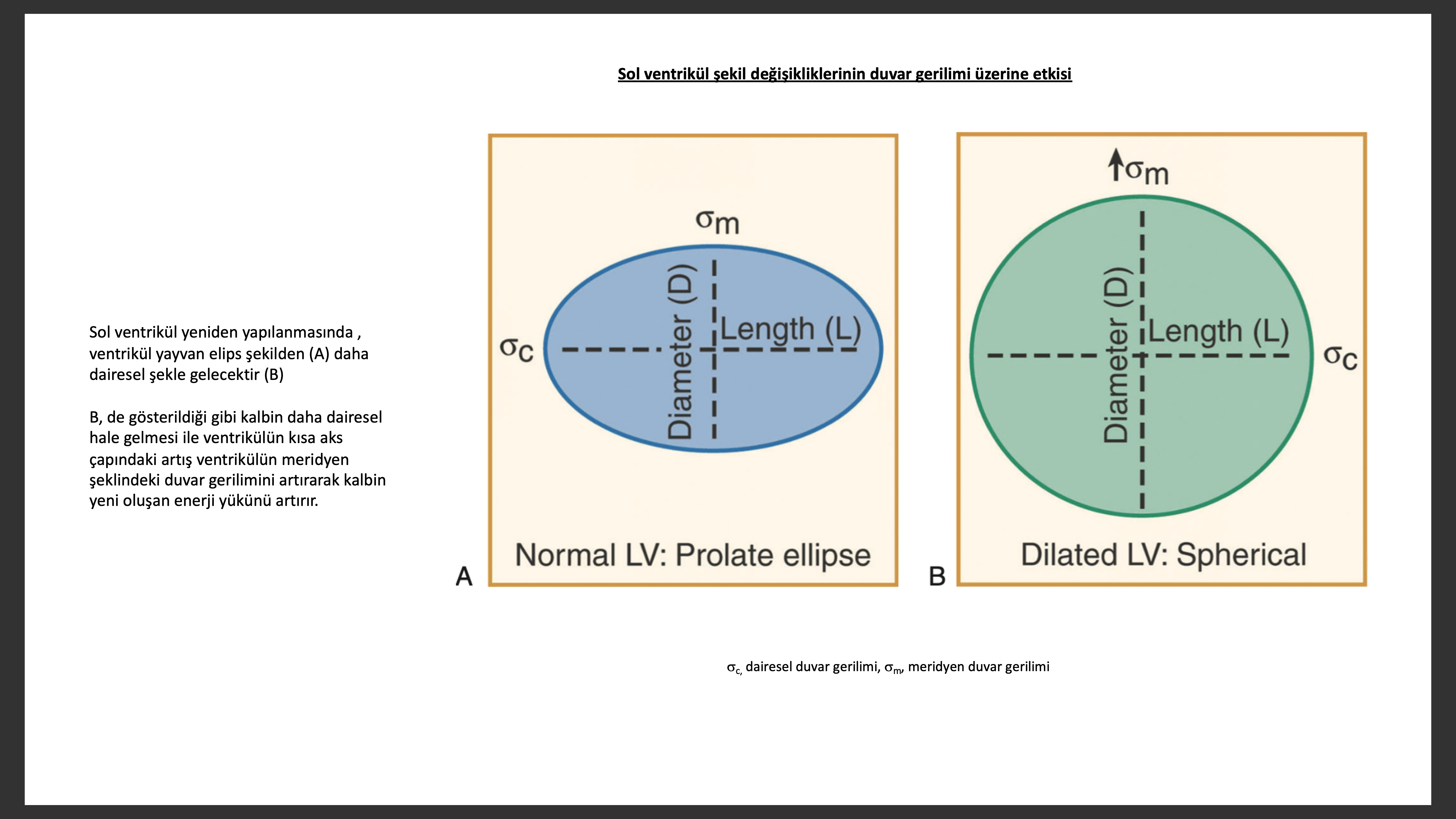 Yeniden yapılanan ventrikül anormal geometrisi hakkındaki ilk gözlem yeniden yapılanan kalbin sadece büyük değil aynı zamanda daha küresel olduğu şeklindedir. Bu noktada önemli olan sol ventrikül şeklinin yayvan elips şeklinden dairesel hale dönmesinin sol ventrikülün meridyen şeklinde duvar gerilimini artırması yetmezlikli kalpte yeni yapılanan enerji yükü meydana getirir. Diyastol sonu ventrikül yükü, sistol başındaki ventriküle art yük olarak eklendiği için, sol ventrikül dilatasyonu kendi başına mekanik enerji harcamasını artırarak yetmezlik olan kalpteki enerji harcama problemini artırır.
Yeniden yapılanan ventrikül anormal geometrisi hakkındaki ilk gözlem yeniden yapılanan kalbin sadece büyük değil aynı zamanda daha küresel olduğu şeklindedir. Bu noktada önemli olan sol ventrikül şeklinin yayvan elips şeklinden dairesel hale dönmesinin sol ventrikülün meridyen şeklinde duvar gerilimini artırması yetmezlikli kalpte yeni yapılanan enerji yükü meydana getirir. Diyastol sonu ventrikül yükü, sistol başındaki ventriküle art yük olarak eklendiği için, sol ventrikül dilatasyonu kendi başına mekanik enerji harcamasını artırarak yetmezlik olan kalpteki enerji harcama problemini artırır.
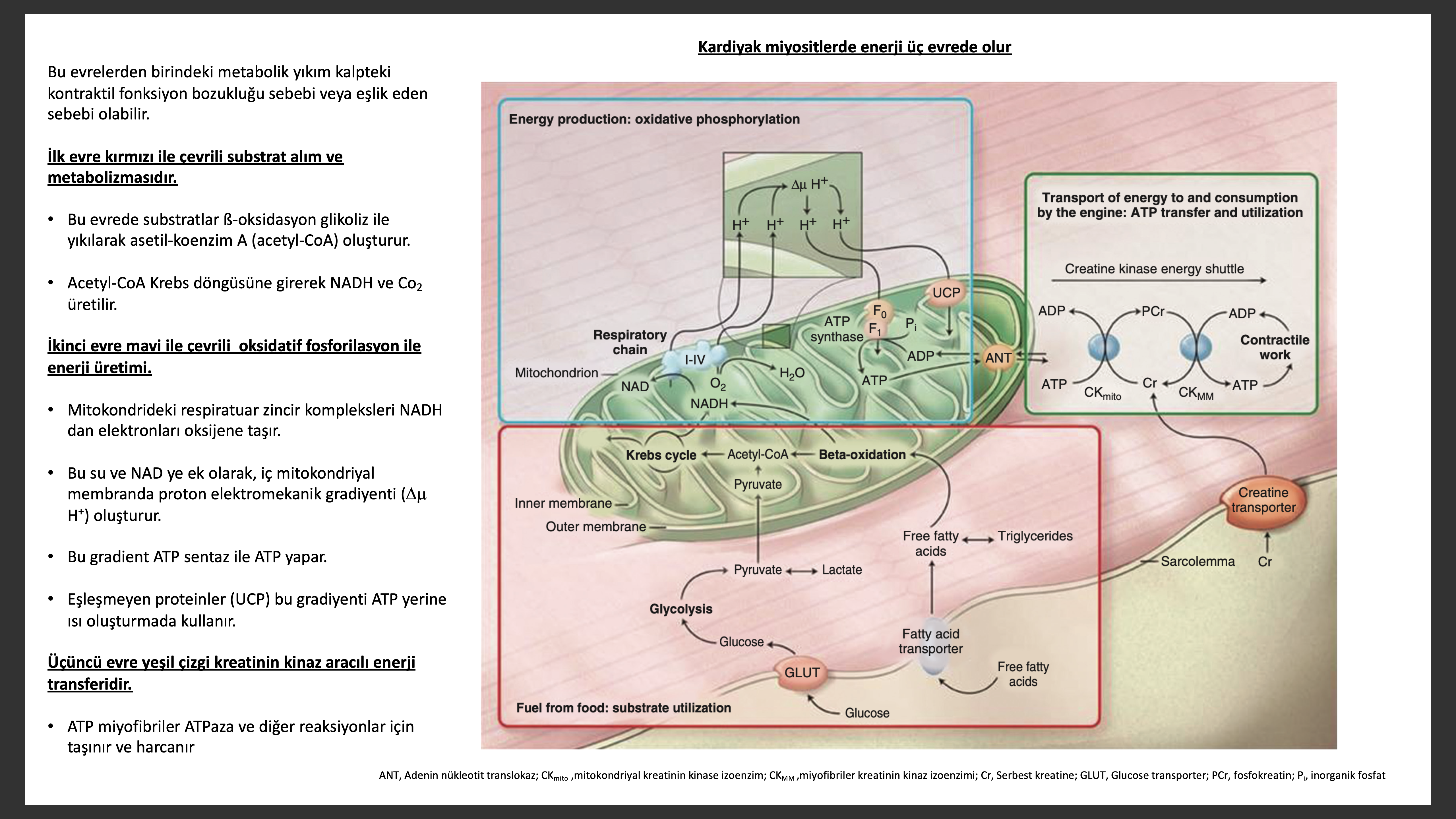
Kardiyak Enerji ve Mitokondri Biyolojisi
Kardiyak miyositde enerji transferi üç evrede gerçekleşir; alım ve metabolizma, oksidatif fosforilasyon ile enerji üretimi ve kreatinin kinaz (CK) aracılı enerji transferi. Bu mekanizma herhangi evresindeki bozukluk kalbin kontraktil bozukluğuna sebep olabilir. Son evre kardiyomiyopati hastalarında yapılan çalışmalarda miyokardiyal ATP yoğunluğu, total adenin nükleotid havuzu (ATP,ADP ve AMP), CK aktivitesi (ATP sentezi için gerekli olan), kreatinin fosfat (CrP) yoğunluğu ve CrP/ATP oranlarını azaltır. Ek olarak azalmış kreatinin fosfokinaz seviyeleri fosfokreatinin taşınmasını yavaşlatır ve kalpteki enerji sarfiyatını artırır. Yine yetersiz kalpte kardiyak enerji sistemlerinin anahtar parçaları azalmıştır. Bugün için bu enerji değişiklikleri sol ventrikül fonksiyon bozukluğu için belirteç mi yoksa sebep mi belli değildir.
Kalp yetersizliğinde düşen ATP içeriğini açıklamak için pek çok mekanizma ileri sürülmüş ancak kaynak kullanımı ile ilgili yalnız bir tanesi kabul görmüştür. Normal koşullarda, yetişkin kalbi enerjisinin çoğunu mitokondride yağ asitlerinin oksidasyonu ile sağlar. Bu önemli enerji metabolik yolunda görev alan genler, özellikle yağ asit-aktive peroksisome proliferator-aktive reseptörleri (PPARs), nükleer reseptör koaktivatörü ve PPAR-gamma coaktivator-1a (PGC-1a) gibi nükleer reseptör süper aile üyeleri ile transkripsiyon yolu ile düzenlenir. Deneysel kalp yetersizliği modellerinde yağ asiti- metabolize eden genlerin baskılanmasına bağlı başlangıçta yağ asit oksidasyonunda azalma ve sonuçta glikolitik metabolizmaya doğru bir kayış gözlenir. Bu gözlemler metabolik geçişin kalp yetersizliğinde faydalı olabileceğini bizlere düşündürdü. Bu metabolik geçişe ek olarak faz II çalışmalarda EF normal ve düşük, kalp yetmezlik hastalarında mitokondiyal ATP sentezini artıran mitokondri-hedef proteini (elamipretide [MTP-13]) çalışmaları yapılmaktadır.
Kalp yetersizliğinde ATP yapılmasındaki azalma mitokondri dinamik bozukluklarından kaynaklana bilir. Maya çalışmaları normal mitokondri morfoloji ve fonksiyonunun korunmasının “mitokondri dinamisi” denilen mitokondriyal füzyon ve fizyonun (bölünme) birlikte çalışması ile olan dinamik dengesinin korunması ile olabileceğini göstermiştir. Mitokondriyal füzyon ve fizyon dengesi kalpteki mitokondri sayı, morfoloji ve aktivitesini belirler. Füzyon ve fizyon, enerjiden ROS üretimine, Ca2+ hemostazına ve hücre ölümüne kadar pek çok mitokondrial fonksiyonu düzenler. Kalp yetersizliğinde çalışmalar sınırlı olsa da eldeki bilgiler füzyonun azalıp, oksijen harcanması azalma ve mitokondri metabolizma değişikliği yönündedir. Mitokondri dinami anormallikleri apoptotik ve otofajik hücre sinyal yolakları üzerinden hücre ölümüne katkıda buluna bilir. Son dönem DCM, miyokardiyal hibernasyon ve konjenital kalp hastalıklarında anormal olarak ufak ve parçalanmış mitokondrilerin izlenmesi mitokondriyal füzyon/fizyonun kalp hastalarında düzenlenmesinin bozulduğunu düşündürmüştür. Ancak halen mitokondriyal füzyon ve fizyon bozukluğunun kalp yetersizliğinde bir sebep mi yoksa miyokardiyal zedelenme ile eş zamanlı bir sonuç mu olduğunu söylemek halen mümkün değildir.
Ventrikül dilate olması ve yeniden yapılanması ile aynı zamanda sol ventrikül duvarının incelir. Sol ventrikül dilatasyonu ile duvar incelmesi ve art yük artışı fonksiyonel bir “art yük uyuşmazlığı” yaparak ileri doğru kardiyak debiyi azaltır. Sol ventrikül gerilim artışı esneme-aktive genlerin (anjiyotenin II, ET,TNF) baskınlığını artırarak ve esneme etkisi ile hipertrofik sinyal yolağını aktive edebilir. Ek olarak artmış diyastol sonu duvar gerginliği aralıklı olarak subendokardiyum perfüzyonunu bozarak sol ventrikül fonksiyonunu bozar, oksidatif stresi artırır sonuçta serbest radikal oluşmasına duyarlı gen ailelerini aktive eder (ör, TNF, IL-1b). İlerleyici sol ventrikül dilatasyonu ile ortaya çıkan diğer problem ise papiller adeleler arasındaki mesafenin açılarak mitral kapakta kaçağa ve fonksiyonel mitral yetersizliğe sebep olmasıdır. Ek olarak ileri doğru kan akım kaybı, mitral yetmezlikte ventrikülde daha fazla hemodinamik yüklenmeye sebep olur. Sol ventrikül yeniden yapılanması ile oluşan mekanik yük ile sol ventrikül dilatasyonunda artma, ileri doğru kalp debisinde azalma ve hemodinamik yükte artış hastanın nörohormonal durumundan bağımsız olarak sol ventrikül fonksiyonunu bozmak için yeterlidir.
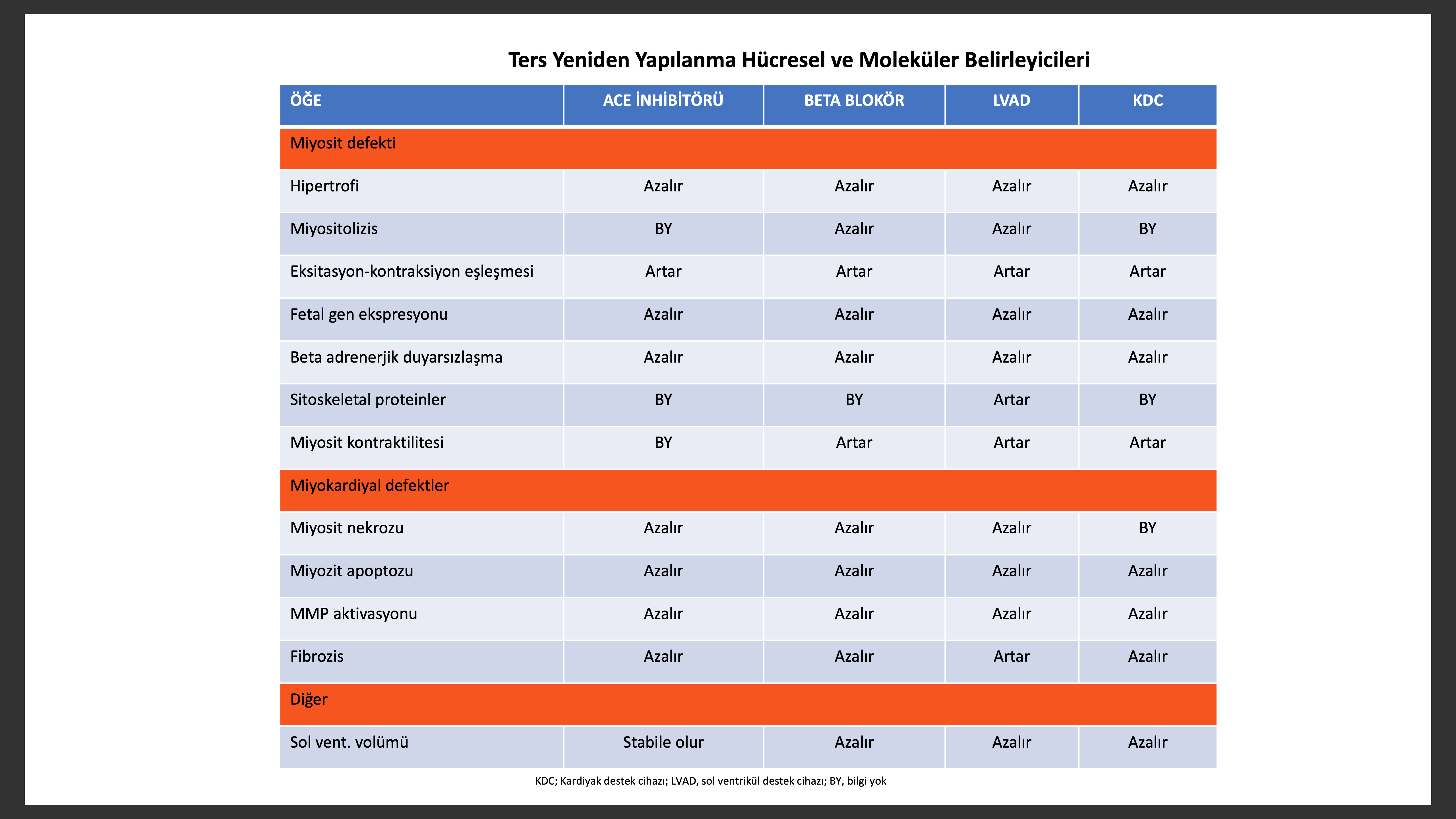
Sol Ventrikül Yeniden Yapılanması Geri Dönüşe Bilirliği
Klinik çalışmalarda kalp yetersizliği morbitite ve mortalitesini azaltan ilaç ve cihazların aynı zamanda sol ventrikül volüm ve kitlesini azaltarak ventrikülü eski dairesel haline getirebileceği gösterilmiştir. Bu yararlı değişiklikler kardiyak miyozit boy ve fonksiyonunda bir seri entegre biyolojik değişikliğin toplamına eşlik eden sol ventrikül yapı ve organizasyonu ile sol ventrikül diyastol sonu basınç-volüm ilişkisini normale doğru gitmesi şeklindedir.
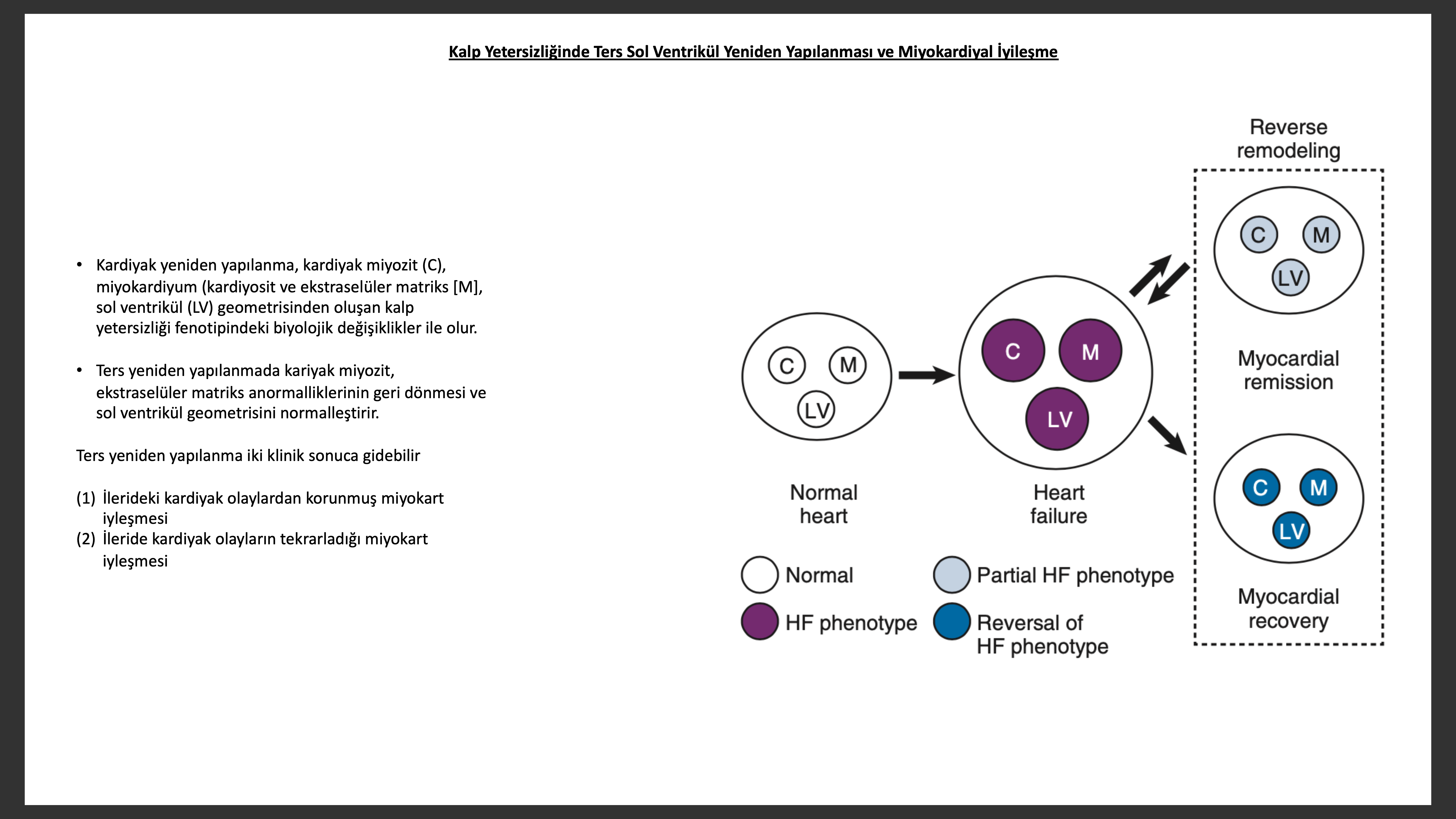 Terminolojik olarak bu değişikliğe ters sol ventrikül yeniden yapılanması (reverse LV remodeling) denir. İlginç olarak bazı hastalarda bu yeniden yapılanmanın düzelmesi spontan olarak olabilmektedir. Spontan veya tedavi ile olsun geri dönüş olan hastalarda klinik kalp yetersizliği hadiseleri daha seyrek olmaktadır. Bu fenomen miyokart iyileşmesi olmaktadır. Her ne kadar miyokart iyileşmesi ve sol ventrikül yeniden yapılanmasının terse dönüşü kalp yetersizliğinin medikal veya cihaz ile fenotipik düzelmesi için benzer terimler olarak kullanılsalar bile literatür bu iki fenomen arasında farklılıklar olduğunu ve bunların sinonim olmadıklarını söylemektedir. Bugün ki anlamı ile ters yeniden yapılanma terimi hücresel, miyokardiyal ve anatomik anormalliklerin yeniden yapılanmış ventrikülde biyolojik olarak geri dönüşünü tarif eder. Kalpleri ters yeniden yapılanma geçiren hastalar ileride potansiyel olarak bir, iki duruma maruz kalabilir: ileride kalp yetersizliği hadiselerinden koruna bilir, veya tekrarlayan kalp yetersizliği atakları olacaktır. Ters yeniden yapılanmada farklı klinik sonuçlanmalardan farklı olarak miyokardiyal iyileşme (myocardial recovery) moleküler, hücresel, miyokardiyal ve sol ventrikül geometrisinde ileride kalp yetersizlik olayları olmayacak şekilde düzelmeyi ifade eder. Miyokardiyal remisyon terimi kardiyak yeniden yapılanma ile olan değişikliklerin normalleşmesi için kullanılır, ancak tam olarak yüklenme veya normal koşullarda tekrarlıyan kalp yetersizliği hadiselerinden korumada yetersizdir. Miyokardiyal iyileşme ile remisyon arasındaki biyolojik farklılıklar tam olarak bilinmemekle birlikte remisyonun artmış geri dönülmez zedelenme olan kalplerde fenotipik kalp yetersizliği dönüşmesi iken, iyileşmenin geri dönülmez zedelenme olmamış kalplerdeki fenotipik kalp yetersizliği düzelmesi olduğu düşünülür.
Terminolojik olarak bu değişikliğe ters sol ventrikül yeniden yapılanması (reverse LV remodeling) denir. İlginç olarak bazı hastalarda bu yeniden yapılanmanın düzelmesi spontan olarak olabilmektedir. Spontan veya tedavi ile olsun geri dönüş olan hastalarda klinik kalp yetersizliği hadiseleri daha seyrek olmaktadır. Bu fenomen miyokart iyileşmesi olmaktadır. Her ne kadar miyokart iyileşmesi ve sol ventrikül yeniden yapılanmasının terse dönüşü kalp yetersizliğinin medikal veya cihaz ile fenotipik düzelmesi için benzer terimler olarak kullanılsalar bile literatür bu iki fenomen arasında farklılıklar olduğunu ve bunların sinonim olmadıklarını söylemektedir. Bugün ki anlamı ile ters yeniden yapılanma terimi hücresel, miyokardiyal ve anatomik anormalliklerin yeniden yapılanmış ventrikülde biyolojik olarak geri dönüşünü tarif eder. Kalpleri ters yeniden yapılanma geçiren hastalar ileride potansiyel olarak bir, iki duruma maruz kalabilir: ileride kalp yetersizliği hadiselerinden koruna bilir, veya tekrarlayan kalp yetersizliği atakları olacaktır. Ters yeniden yapılanmada farklı klinik sonuçlanmalardan farklı olarak miyokardiyal iyileşme (myocardial recovery) moleküler, hücresel, miyokardiyal ve sol ventrikül geometrisinde ileride kalp yetersizlik olayları olmayacak şekilde düzelmeyi ifade eder. Miyokardiyal remisyon terimi kardiyak yeniden yapılanma ile olan değişikliklerin normalleşmesi için kullanılır, ancak tam olarak yüklenme veya normal koşullarda tekrarlıyan kalp yetersizliği hadiselerinden korumada yetersizdir. Miyokardiyal iyileşme ile remisyon arasındaki biyolojik farklılıklar tam olarak bilinmemekle birlikte remisyonun artmış geri dönülmez zedelenme olan kalplerde fenotipik kalp yetersizliği dönüşmesi iken, iyileşmenin geri dönülmez zedelenme olmamış kalplerdeki fenotipik kalp yetersizliği düzelmesi olduğu düşünülür.